Exceptional problems demand exceptional computers
Proteins perform most of the daily work of the cell, they catalyse most of the biochemical reactions, initiate and mediate the electrical signals in our neurons and form an important part of the structure of our bodies. Formed by up to several thousand amino acids (from a set of 20 different types) chained one after the other, proteins fold under the effect of non-covalent forces between different parts of the molecule into shapes of functional significance; shapes that are not static but change constantly, randomly and also react to external influence. These conformational changes have an impact in their function, allowing the proteins to interact with one another or with other signalling molecules (hormones, drugs…). Protein misfolding or modifications of conformational changes lead to illnesses, including Alzheimer’s and Parkinson’s diseases, and much money and effort are being put into understanding protein structures, folding pathways and interactions with other molecules.
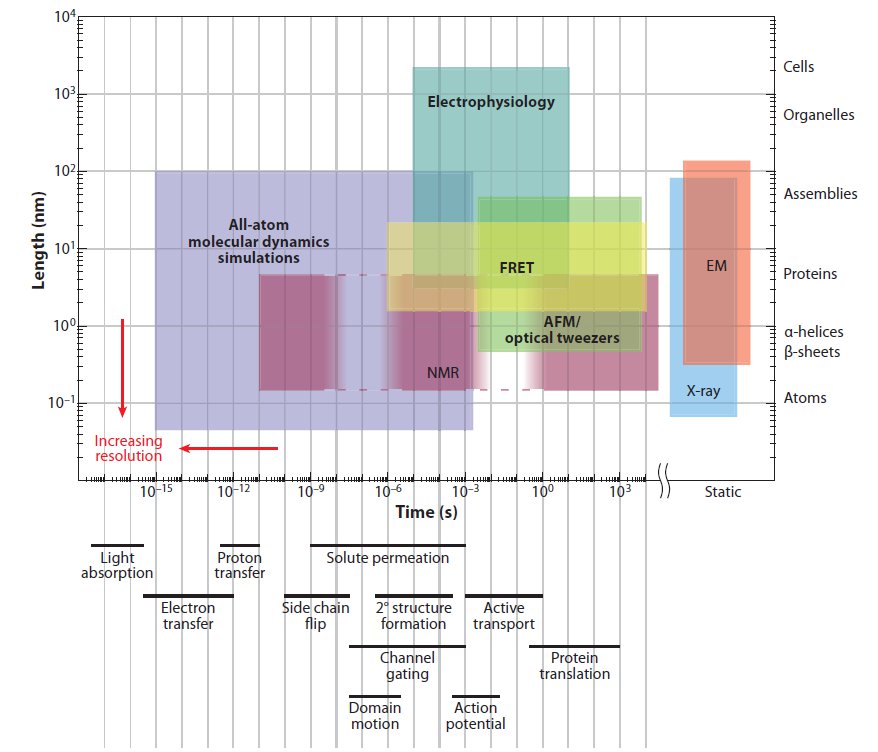
Several experimental tools are used to study protein conformation, but they all are limited in their spatial and temporal resolution (Fig 1). Indeed, the most powerful among them, X-ray diffraction, only provides a snapshot of the structure of proteins. If we want to study protein dynamics (how the conformation of a molecule changes over time) then computer simulation provides a good alternative. An exact simulation will require solving the quantum mechanical equations for large molecules, but this is computationally too complicated to be practical. Molecular dynamics (MD) simulates each atom of the molecule as evolving following the rules of classical mechanics and semi-empirical force fields. MD simulations have been used since the mid-seventies to understand biochemical processes, and currently account for most of the computer time assigned for biomedical research in the supercomputer centres of the National Science Foundation, USA.
Despite its popularity, MD simulations are computationally expensive. The need to capture fast atom vibrations requires computation steps of a few femtoseconds, and each step involves one billion operations for a molecule of one hundred thousand atoms. To simulate one millisecond in the life of a single protein molecule we need around one trillion steps and a sextillion (10^21) operations. As a result, even using the most powerful supercomputers available, most MD simulations only study processes that last nanoseconds or microseconds. It would take an impractically long supercomputer run time to reach the millisecond timescales in which many protein conformational changes and interactions take place. In a recent paper 1, Ron O. Dror and his colleagues from D. E. Shaw Research review recent developments that are overcoming this problem.
One solution, computing different parts of the molecule in parallel, has proven to be particularly difficult. Recent innovations in parallel algorithms have partially improved the situation, allowing supercomputer clusters to compute over the microsecond timescale. Indeed, in some problems,part of the simulation can be divided in many separate short trajectories that can be run in parallel in hundreds of thousands of personal computers. This is the Folding@Home project, that gives anyone with a computer (or even a modern PlayStation) and internet connection the opportunity to donate simulation time and collaborate with scientific research. More than one hundred peer-reviewed papers have been produced thanks to this initiative.
The biggest advances in optimising MD simulation have been made following that route of adapting our tools to the task. This is a well-known solution in engineering and in biology. For example, eyes are adapted to the scenes they are likely to see, making them more efficient, and ears for the sounds they are likely to hear. In computer engineering, graphics processing units (GPUs) were designed for operations with graphics and in that task they outperform much more powerful general processors. They are a particularly good example of the fact that purpose built hardware may lead to more efficient computation of particular problems.
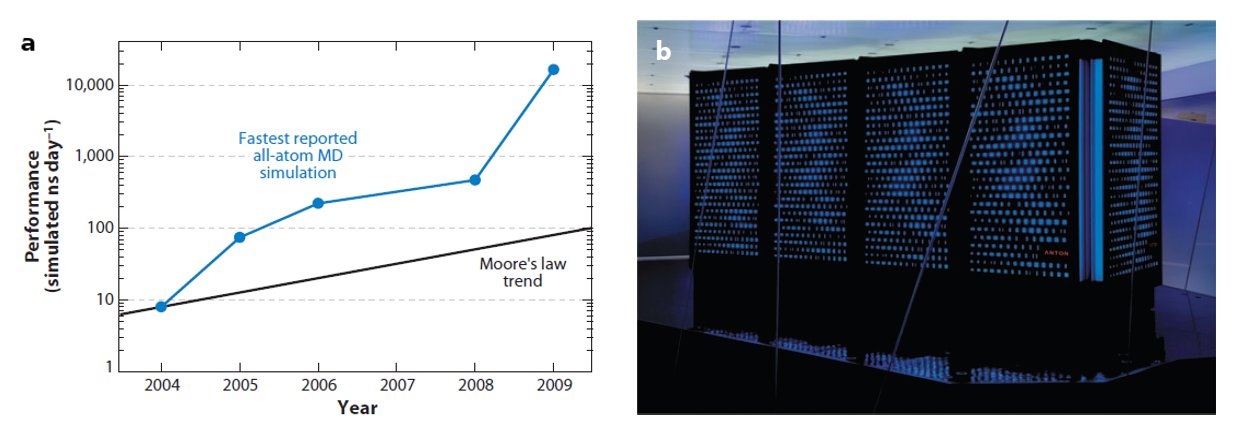
D. E. Shaw Research followed this idea in designing a purpose-designed MD simulation machine, called Anton. Its chips, containing an “array of arithmetic units hard-wired for the computation of particle interactions”, are connected among them in a way that avoids the use of computationally expensive memory management. Anton carries out all its operations in purpose-built circuits, as opposed to previous projects (FASTRUN, MD Engine, MDGRAPE) where only the most computationally expensive parts of the simulation were optimised. Thanks to this special architecture, Anton can perform 20 microseconds of an all-atom MD simulation per day, around 100 times faster than any other alternative.
In their review, Anton developers suggest problems that are likely to benefit from the developments in this field. Better MD simulations will help in the development of drugs, by producing more accurate predictions on how effectively they would bind to proteins, and in the design of proteins to be used as biosensors for cancer or antibodies. Moreover, the study of the dynamics of other large molecules such as RNAs and DNAs will also benefit from the construction of special, purpose-built, computers.
References
- Ron O. Dror, Robert M. Dirks, J.P. Grossman, Huafeng Xu and David E. Shaw, “Biomolecular Simulation: A Computational Microscope for Molecular Biology”, Annual Review of Biophysics 41: 429-452 (2012) DOI: http://dx.doi.org/10.1146/annurev-biophys-042910-155245 ↩
3 comments
You are lucky in the classical world. In quantum systems very simple problems demand exceptional computers. As an example, simulating a general (mixed) system composed by 7 qubits (two level systems) is quite hard with a laptop. More qubits require a lot of RAM memory, and beyond 12 is almost impossible.
But take into account that proteins are not fully classical either. What if a proton tunnels between two residues and this affects the folding process? Maybe it is not common, but I don’t see right now why it could not happen.
And of course, when delocalized electrons enter the scene (e.g. photosynthyesis), quantum coherence can be a critical factor and classical simulations are not going to work.
[…] Exceptional problems demand exceptional computers en Mapping […]