The anisotropic behaviour of ultrafast electron transfer at the metal/organic interface
The anisotropic behaviour of ultrafast electron transfer at the metal/organic interface
Quantum objects are not abstract entities. On the contrary, they are quite real, As such, individual quantum objects can be detected, manipulated and their characteristics analysed using the appropriate techniques.
Take the case of a single molecule adsorbed on a substrate. Scanning Tunneling Microscopy (STM) would reveal electron densities of molecular orbitals, could be used to control the charge state of the adsorbate, or to visualize bond formation between atoms and organic molecules. Energies and lifetimes of decaying quasi-stationary electronic states of individual molecules and nanostructures adsorbed on metal surfaces can be determined with Scanning Tunneling Spectroscopy (STS);
this information is nicely completed by Time-Resolved Two-Photon-Photoemission (TR-2PPE) studies of the dynamics of excited states at pristine and adsorbate coated surfaces.
But if the substrate is a metal, a serious challenge appears: the time the processes need to take place is reduced to femtoseconds.
For molecules adsorbed at metal surfaces, the coupling between the adsorbate-localized electronic states and the continuum of electronic states of the metal leads to the de-excitation of the molecule-localized excited state via one-electron energy-conserving transfer from the molecular orbital into the metal (this is called resonant electron transfer, RET), or via multi-electron relaxation. Usually these processes are extremely efficient resulting in an extremely short lifetime of the excited state. This femtosecond-scale lifetime harms the luminescence or surface reactivity, which evolve at much longer characteristic times, on the picosecond and even nanosecond timescales.
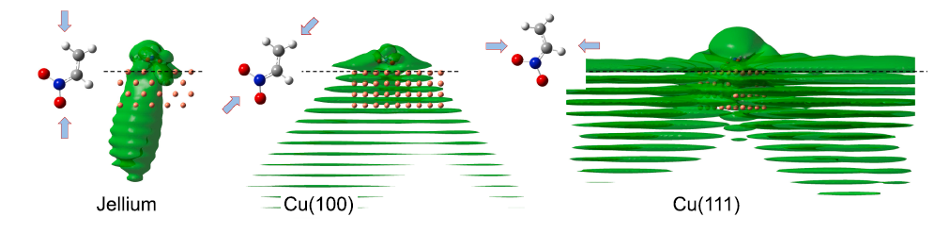
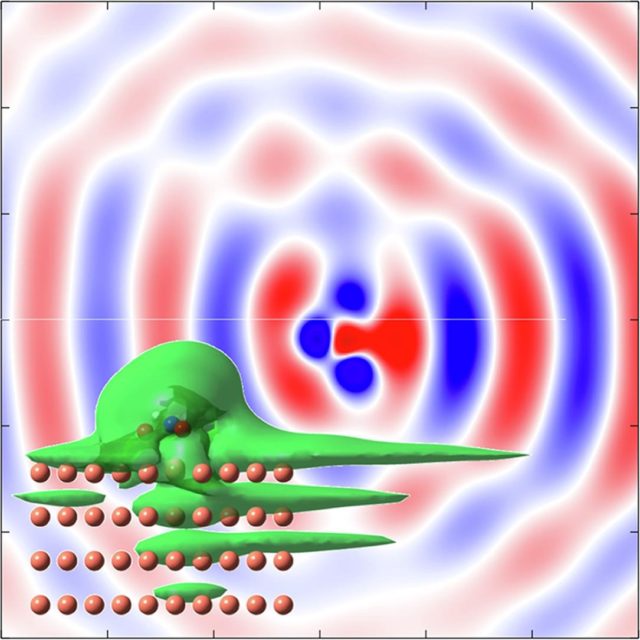
To understand RET, the most efficient mechanism because of its one-electron nature, is of particular importance for comprehending how molecules self-assembly at metal surfaces. To this end, a team of researchers has chosen 1 nitroethylene interacting with a free electron metal, Cu(100), and Cu(111) surfaces, characterized by their very different electronic structure, to model the ultrafast dynamics of RET from an adsorbed molecular anion into a metal substrate. The researchers find that a strong coupling with the metal surface turns the molecule-localized π* orbital into a resonance with femtosecond lifetime, and that many characteristics of the RET can be understood as resulting from the surface projected band structure.
This may sound familiar or, at least, not at all unexpected, but the scientists demonstrate that the decay of the molecule-localized resonance is a much more complex process. The arrangement of the atoms within the molecule, and the electronic properties of the molecule–metal interface lead to specific decay characteristics, importantly, with a pronounced anisotropy of the hot electron distribution in the metal resulting from the decay of the molecular π* resonance.
This finding is of particular relevance for such an active area of surface science as surface-supported organic networks. The strong anisotropy in the adsorbate/ substrate electronic coupling is of importance for their electronic and structural properties, which depend on indirect adsorbate–adsorbate interactions with the substrate. The results are also of interest for engineering the surface electronic structure, where the electronic surface state of the substrate is confined using organic adsorbates, and open a way to a rational design of hybrid metal/organic interfaces with tailored electronic properties.
Author: César Tomé López is a science writer and the editor of Mapping Ignorance
Disclaimer: Parts of this article may have been copied verbatim or almost verbatim from the referenced research papers.
References
- Fernando Aguilar-Galindo, Andrey G. Borisov & Sergio Díaz-Tendero (2021) Unveiling the anisotropic behaviour of ultrafast electron transfer at the metal/organic interface Applied Surface Science doi: 10.1016/j.apsusc.2021.149311 ↩