CASZ1 promotes T-cell acute lymphoblastic leukemia
CASZ1 promotes T-cell acute lymphoblastic leukemia
T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive hematological malignancy that results from transformation and clonal expansion of developmentally-arrested T-cell progenitors 1. Conventional risk-adjusted multi-agent chemotherapy allows for high 5-year event-free survival rates in children. However, a significant number of patients still relapse or do not respond to therapy. Consequently, there have been considerable efforts to better understand the cell-intrinsic lesions and micro-environmental underpinnings of the disease, resulting in the identification of numerous key genetic 23 and cell-extrinsic 45 alterations involved in the development and resistance to treatment of T-ALL. Among these, several candidates have revealed potential to translate into novel, hopefully less toxic and more efficient therapies 6, which may contribute to circumventing resistance to chemotherapy. Thus, identification of new molecular regulators of T-ALL should contribute to a better understanding of the disease and, consequently, to the improvement of therapeutic approaches.
CASZ1 is a highly conserved 7 zinc finger transcription factor essential for blood vessel assembly 8, cardiomyocyte differentiation and proliferation 9 and heart morphogenesis 10. Interestingly, the involvement of CASZ1 in cancer varies depending on the type of tissue and it has not been defined yet its role in T-ALL. In the present paper, Bruno A. Cardoso and collaborators demonstrate that CASZ1b has an oncogenic role in T-ALL mediated by PI3K-AKT-mTOR signaling activation11.
The role of CASZ1 in T-ALL
To analyze the role of CASZ1 in T-ALL, the authors firstly analyzed CASZ1 expression in leukemia samples collected from T-ALL patients as compared to normal thymocytes subsets representative of the main T-cell developmental. Thus, they found that CASZ1b (the short isoform of CASZ1) was significantly up-regulated in T-ALL cells. Despite CASZ1 levels were generally increased in T-ALL as compared to normal T-cell precursors, they noticed that high expression of TAL1 (a transcription factor that is aberrantly expressed in a high percentage of patients with T-ALL) expression was directly correlated with higher levels of CASZ1 and few expression of TAL1 correlated with low CASZ1 levels. These results suggested a possible association between TAL1 and CASZ1 expression in T-ALL patients. Thus, the authors decided to evaluate whether TAL1 might transcriptionally regulate CASZ1. To understand the role of TAL1, they forced its expression in two TAL1-negative T-ALL cell lines. They found that TAL1 overexpression led to the upregulation of CASZ1 expression, whereas TAL1 inactivation in TAL1-positive cells downregulated CASZ1, reinforcing the positive regulatory link between TAL1 and CASZ1 in T-ALL.
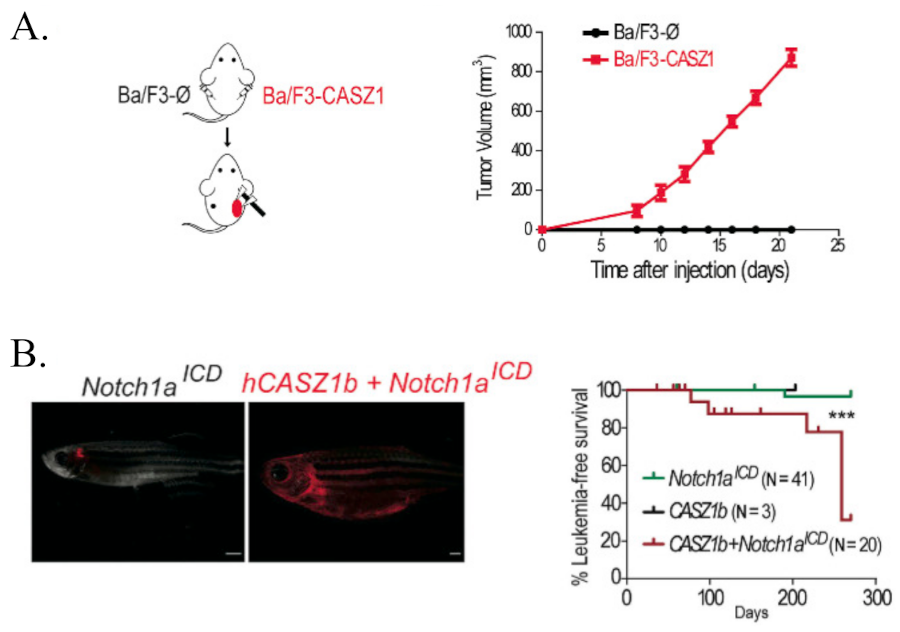
Given that CASZ1 is overexpressed in T-ALL cells, authors postulated that CASZ1 should have an oncogenic role in lymphoid cells. To test this possibility, they expressed CASZ1 in a stable manner in IL-3-dependent Ba/F3 cells (a murine interleukin-3 dependent pro-B cell line). Interestingly, Ba/F3 cells expressing CASZ1 in the absence of IL-3 maintained their viability and proliferated throughout time to similar levels as control cells cultured with the growth factor, supporting the oncogenic role of this gene. Next, to identify potential mechanisms by which CASZ1 exerted its oncogenic effects, they analyzed the transcriptional program engaged by CASZ1 in Ba/F3 cells and showed that CASZ1 affected the expression of a high number of genes. Remarkably, gene analysis indicated that PI3K-AKT signaling was highly enriched in CASZ1 overexpressing cells and indeed, this overexpression upregulated the phosphorylation levels of members of the PI3K-AKT-mTOR signaling pathway, such as AKT, mTOR and S6. To test the involvement of these signaling pathways in CASZ1-mediated transformation, they treated CASZ1-expressing Ba/F3 cells with two distinct PI3K/mTOR inhibitors. As expected, both inhibitors reduced PI3K-AKT-mTOR signaling, and abrogated the effects of CASZ1 on viability and proliferation. After these stricking results, they decided to evaluate whether, similar to Ba/F3 cells, CASZ1-mediated effects in T-ALL cells involved upregulation of PI3K-AKT-mTOR signaling. They compared the expression levels of all the transcripts to those of CASZ1 in a T-ALL dataset and the results showed that genes whose expression correlates with that of CASZ1 were enriched in different pathways, including PI3K-AKT signaling. Besides, immunoblot analysis confirmed that T-ALL cell lines overexpressing CASZ1 showed higher levels of PI3K-AKT-mTOR signaling activation than controls.
Next, they evaluated whether CASZ1 was able to promote tumorigenesis in vivo. To demonstrate that, they subcutaneously grafted empty vector- and CASZ1-transduced Ba/F3 cells into opposite flanks of immunosuppressed NOD/SCID mouse. Whereas none of the control transplants originated any detectable tumors, 10 out of 10 CASZ1-expressing transplants originated large tumor masses within 20 days since inoculation. To ascertain whether PI3K-AKT-mTOR signaling pathway was essential for the maintenance of CASZ1-triggered tumors in vivo, mice were randomly divided into two cohorts that either received vehicle or a PI3K/mTOR inhibitor. Interestingly, treatment with the inhibitor clearly delayed tumor growth. To determine whether CASZ1 promotes tumorigenesis in the context of T-ALL, they next tested whether CASZ1 accelerates NOTCH-induced T-cell leukemia in zebrafish. They generated mosaic zebrafish lines expressing intracellular NOTCH1 (ICN1, which translocate to the nucleus and executes functions by turning on the transcription of target genes) and/or CASZ1. Although with long latency, they found that CASZ1 leaded to leukemia/lymphoma development in 6 of 20 animals expressing CASZ1 and ICN1, as opposed to only 1 of 41 animals expressing ICN1 alone that developed leukemia. Finally, malignant cells in CASZ1+ICN1 zebrafish showed a reduction of cell apoptosis compared to control cells in ICN1 zebrafish.
Finally, to investigate the biological impact of CASZ1 in human T-ALL, they tried to silence CASZ1 in T-ALL cell lines. Since the silencing efficiency was too poor, they decided to express ectopically CASZ1 in several T-ALL cell lines. Unfortunately they did not observed any change in T-ALL cell viability or proliferation. Given that basal viability and growth rate of T-ALL cell lines in regular culture conditions are high, these results may mask positive effects of some oncogenes. Similar to TAL1, CASZ1 overexpression protected T-ALL cells from serum starvation induced-apoptosis and conferred resistance to treatment with the chemotherapeutic drugs currently used to treat patients with T-ALL. Based on these findings, they hypothesized that high expression of CASZ1 might correlate with unfavorable outcome. To corroborate that, they focused in a cohort comprising exclusively of relapse T-ALL patients with high CASZ1 levels and found that was associated with particularly poor prognosis, as the CASZ1-high group displayed faster progression to disease relapse and decreased overall survival. Interestingly, in agreement with their findings that TAL1 regulates CASZ1 in T-ALL, TAL1 was also associated with poor prognosis in this cohort of patients.
In summary, the authors demonstrated that CASZ1 is upregulated in T-ALL, protecting T-ALL cells from stress-induced apoptosis and inducing the activation of the PI3K-AKT-mTOR signaling pathway, which is required for transformation in vitro and tumor growth in vivo.
Author: Marta Irigoyen is a postdoctoral researcher at CIC bioGUNE
References
- Belver L, Ferrando A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer 2016; 16: 494-507 PMID: 27451956 DOI: 10.1038/nrc.2016.63. ↩
- Weng AP, Ferrando AA, Lee W, Morris JP 4th, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004; 306: 269-271. PMID: 15472075 DOI: 10.1126/science.1102160. ↩
- Gutierrez A, Sanda T, Grebliunaite R, Carracedo A, Salmena L, Ahn Y, Dahlberg S, Neuberg D et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood2009; 114: 647-650. PMID: 19458356 DOI: 10.1182/blood-2009-02-206722. ↩
- Silva A, Laranjeira AB, Martins LR, Cardoso BA, Demengeot J, Yunes JA, Seddon B, Barata JT. IL-7 contributes to the progression of human T-cell acute lymphoblastic leukemias. Cancer Res 2011; 71: 4780-4789. PMID: 21593192 DOI: 10.1158/0008-5472.CAN-10-3606. ↩
- Pitt LA, Tikhonova AN, Hu H, Trimarchi T, King B, Gong Y, Sanchez-Martin M, Tsirigos A et al. CXCL12-producing vascular endothelial niches control acute T cell leukemia maintenance. Cancer Cell 2015; 27: 755-768. PMID: 26058075 DOI: 10.1016/j.ccell.2015.05.002. PMID: 26058075. ↩
- Peirs S, Matthijssens F, Goossens S, Van de Walle I, Ruggero K, de Bock CE, Degryse S, Canté-Barrett K et al. ABT-199 mediated inhibition of BCL-2 as a novel therapeutic strategy in T-cell acute lymphoblastic leukemia. Blood 2014; 124: 3738-3747. PMID: 25301704 DOI: 10.1182/blood-2014-05-574566. ↩
- Liu Z, Yang X, Tan F, Cullion K, Thiele CJ. Molecular cloning and characterization of human Castor, a novel human gene upregulated during cell differentiation. Biochem Biophys Res Commun 2006; 344: 834-844 PMID: 16631614 DOI: 10.1016/j.bbrc.2006.03.207. ↩
- Charpentier MS, Christine KS, Amin NM, Dorr KM, Kushner EJ, Bautch VL, Taylor JM, Conlon FL. CASZ1 promotes vascular assembly and morphogenesis through the direct regulation of an EGFL7/RhoA-mediated pathway. Dev Cell 2013; 25: 132-143. PMID: 23639441 DOI: 10.1016/j.devcel.2013.03.003. ↩
- Dorr KM, Amin NM, Kuchenbrod LM, Labiner H, Charpentier MS, Pevny LH, Wessels A, Conlon FL. Casz1 is required for cardiomyocyte G1-to-S phase progression during mammalian cardiac development. Development 2015; 142: 2037-2047. PMID: 25953344 DOI: 10.1242/dev.119107. ↩
- Huang RT, Xue S, Wang J, Gu JY, Xu JH, Li YJ, Li N, Yang XX et al. CASZ1 loss-of-function mutation associated with congenital heart disease. Gene 2016; 595: 62-68. PMID: 27693370 DOI: 10.1016/j.gene.2016.09.044. ↩
- Cardoso BA, Duque M, Gírio A, Fragoso R, Oliveira ML, Allen JR, Martins LR, Correia NC et al. CASZ1 upregulates PI3K-AKT-mTOR signaling and promotes T-cell acute lymphoblastic leukemia. Haematologica 2024; 109: 1713-1725. PMID: 38058200 DOI: 10.3324/haematol.2023.282854. ↩