TRAF6 functions as a tumor suppressor in myeloid malignancies
TRAF6 functions as a tumor suppressor in myeloid malignancies
Hematopoietic stem cells (HSC) accumulate somatic mutations during ageing in healthy individuals 1. Despite most of these mutations are inconsequential, some HSC can acquire specific mutations providing a competitive advantage leading to a process known as “clonal hematopoiesis of indeterminant potential” (CHIP) 2. The most common CHIP mutations affects to DNMT3A and TET2 genes 3 and are a strong risk factor for subsequent hematologic cancer (3). However, leukemia occurs with low frequency 45. These findings indicate that additional alterations are needed for malignant transformation of pre-leukemic hematopoietic stem and progenitor cells (HSPCs). Thus, the authors of the present work aimed to elucidate the factors – beyond mutated genes – that cooperate to induce myeloid malignancies with CHIP-associated pre-leukemic mutations 6.
To identify cooperating events in pre-leukemic HSCs, the authors used the Tet2 mutation. Firstly, they performed an in vivo RNAi screen using HSPCs devoided of Tet2 (Tet2−/− HSPCs). Because the dysregulation of ubiquitination and proteosomal processes are frequently associated with blood cancer 7, they utilized a direct shRNA library targeting its regulators. Therefore, they transduce Tet2−/− HSPCs with shRNA of genes involved in these processes and grafted them into lethally-irradiated host mice. Since these mice showed signs of myeloid disease, they used deep sequencing to readout the relative representation of shRNA in the Tet2−/− HSPCs pre-transplantation and in the primary leukemic cells isolated from sick mice. Interestingly, a small fraction of the shRNAs was represented in the primary leukemic cells collected from the sick mice and even less showed >5-fold average enrichment. Next, they ranked the shRNAs based on their higher enrichment and they observed that genes targeted by the top ranked shRNAs were involved in different cellular processes. Interestingly, bioinformatic pathway analysis revealed an enrichment of the TLR pathway, mainly due to the presence of the E3 ubiquitin ligase “Traf6”. Thus, they hypothesized that TRAF6 loss-of-function may cooperate with TET2 mutations in the development of myeloid malignancies.
To determine the consequences of TRAF6 deletion on pre-leukemic cells, they used Traf6f/f and Tet2f/f mice (f = “floxed”, it refers to mice with alleles that contain a target gene flanked by specific sequences, that can lead to its deletion in presence of a CRE recombinase enzyme) (5). After serial transplantation assays, they observed that: 1) with the 3rd replating, wild-type (WT) and Traf6−/− cells were unable to form colonies, 2) Tet2−/− cells formed colonies beyond the 5th replating and 3) Traf6−/−;Tet2−/− cells exhibited an increased colony replating potential as compared to Tet2−/− cells. They next performed in vivo competitive repopulation assays. Following engraftment, mice were treated to induce the excision of the floxed alleles (which causes the loss of the Tet2 and Traf6 genes) and then analyzed every 4 weeks for hematopoietic reconstitution. Supporting the previous results, deletion of Tet2 restored the repopulating potential defect of Traf6-deficient cells and contributed to a significant expansion of donor-derived myeloid cells in primary and secondary (bone marrow) BM transplants. So, they next evaluated the effect of concurrent deletion of Traf6 and Tet2 on the development of hematological malignancies performing transplantation of BM cells into WT mice and then monitored for hematological disease. Interestingly, a subset of experimental animals reconstituted with Tet2−/− BM cells developed a fatal myeloid disease after a long latency and all mice reconstituted with Traf6−/−;Tet2−/− BM cells developed a fully penetrant, fatal, and transplantable disease which developed a myeloid leukemia 8.
Since reduced expression of TRAF6 was able to promote a myeloid leukemia in mouse models, they next wanted to determine whether genetic alterations targeting TRAF6 are observed in human myeloid malignancies. Upon inspection of publicly available data, they observed that genetic alterations affecting TRAF6 are present in AML. Thus, they evaluated TRAF6 mRNA expression in AML and correlated that TRAF6 expression occurs in ~30% of AML patients as compared to healthy controls. Moreover, TRAF6 protein was significantly reduced in approximately half of the AML examined patients. To establish the basis for reduced TRAF6 expression in AML, they found significant hypermethylation of CpG sites proximal to the TRAF6 promoter as compared to controls. Moreover, they observed that the majority of AML samples with low levels of TRAF6 displayed aberrant processing of the 5’-end of the TRAF6 transcript. Using the BEAT-AML data set 9 on AML patients stratified on TRAF6 expression (BEAT-AML has an extensive genomic, drug response, and clinical dataset on AML), they showed TET2 mutations in AML patients with low expression of TRAF6.
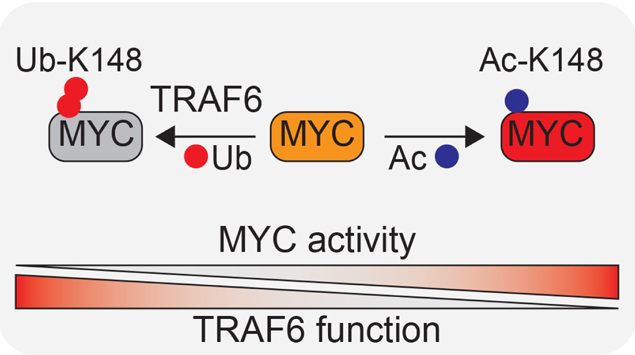
TRAF6 is an E3 ligase that catalyzes the formation of Lysine (K) 63-linked ubiquitin (Ub) chains and its loss represents a secondary disease-modifying alteration that contributes to the transformation of pre-leukemic cells. Knowing that, the authors sought to focus on the mechanistic consequences resulting from loss of TRAF6 and performed a global quantitative Ub capture proteomic in cells devoided of TRAF6 compared to control cells. Of the top ranked candidates, TRAF6 knockdown resulted in reduced ubiquitination of MYC at K148, a post-translational modification implicated in MYC function 10. To test the ability of TRAF6 to directly ubiquitinate MYC, they utilized recombinant proteins in a cell-free biochemical assay and observed that MYC was readily ubquitinated in the presence of TRAF6. Next, they investigated whether ubiquitination of MYC at K148 by TRAF6 affects MYC transcription factor activity observing that TRAF6 overexpression repressed MYC-dependent activity. These results suggested that TRAF6 is able to repress MYC transcriptional activity via ubiquitination of K148.
To map the post-translational modifications on MYC relevant to human AML, the authors immunoprecipitated MYC from AML cells and performed mass spectroscopy. Thus, they observed that both acetylation and ubiquitination modifications were found on K148 in AML. To determine the impact of TRAF6-mediated ubiquitination on MYC post-translational modifications, they first examined the phosphorylation status and total levels of MYC. Total and phosphorylated MYC levels were unchanged in Traf6-deficient HSPCs, suggesting that K148 modifications did not affected MYC levels or its phosphorylation. Although the functional relevance of K148 acetylation on K148 is unclear, such a post-translational modification of MYC could have pleotropic effects. Thus, they postulated that TRAF6 ubiquitination of MYC at K148 prevents its acetylation, which results in diminished MYC activity. Immunoblotting of Traf6−/−;Tet2−/− or Tet2−/− BM progenitors with an Ac-K148-specific MYC antibody revealed that MYC exhibited a reduced Ub-K148 with an Ac-K148 increase upon deletion of Traf6. Knockdown of TRAF6 in human AML cells resulted in increased MYCK148 acetylation, which correlated with loss of MYC ubiquitination. They also examined human AML cell lines and primary AML samples to correlate TRAF6 protein expression and MYCK148 acetylation. As a general trend, they found that both TRAF6Low AML cell lines, and patient samples, had an increased acetylation of MYCK148. These findings revealed that TRAF6 was able to ubiquitinate MYCK148 (which prevented the acetylation on the same residues). In contrast, reduction of TRAF6-mediated ubiquitination of MYCK148 was able to allow the acetylation on the free lysine residue.
To investigate the effects of acetylated K148 on MYC activity, they generated gene-edited cells at K148 to glutamine (K148Q) and corroborated that loss of MYCK148 acetylation resulted in a diminished transcriptional activity in AML. Finally, to determine the consequences of K148 acetylation on MYC function on AML, they also expressed MYC(K148Q) or WT MYC in Tet2−/− BM cells. Transduced cells were grafted in mice showing that the overexpression of MYC(K148Q) in Tet2−/− HSPCs resulted in an increased myeloid blasts and splenomegaly after 8 weeks post transplantation (compared to Tet2−/− HSPCs expressing WT MYC). As such, loss of TRAF6 and a corresponding increase in MYCK148 acetylation results in enhanced MYC-induced oncogenic function and development of AML.
In summary, authors demonstrated that loss of TRAF6 in pre-leukemic cells results in overt myeloid leukemia and is associated with MYC-dependent stem cell signatures. Besides, TRAF6 is repressed in a subset of patients with myeloid malignancies, suggesting that subversion of TRAF6 signaling can lead to acute leukemia.
Author: Marta Irigoyen is a postdoctoral researcher at CIC bioGUNE
References
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli1. N et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415-21. PMID: 23945592 DOI: 10.1038/nature12477. ↩
- Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264-78. PMID: 22817890 DOI: 10.1016/j.cell.2012.06.023. ↩
- Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477-87. PMID: 25426838 DOI: 10.1056/NEJMoa1409405. ↩
- Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, Bock C, Vasanthakumar et al. ADnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2011;44:23-31. PMID: 22138693 DOI: 10.1038/ng.1009. ↩
- Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasanthakumar A et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11-24. PMID: 21723200 DOI: 10.1016/j.ccr.2011.06.001. ↩
- Muto T, Guillamot M, Yeung J, Fang J, Bennett J, Nadorp B, Lasry A, Redondo LZ et al. TRAF6 functions as a tumor suppressor in myeloid malignancies by directly targeting MYC oncogenic activity. Cell Stem Cell. 2022;29:298-314.e9. PMID: 35045331 DOI: 10.1016/j.stem.2021.12.007. ↩
- Narayan S, Bader GD, Reimand J. Frequent mutations in acetylation and ubiquitination sites suggest novel driver mechanisms of cancer. Genome Med. 2016;8:55. PMID: 27175787 DOI: 10.1186/s13073-016-0311-2. ↩
- Kogan SC, Ward JM, Anver MR, Berman JJ, Brayton C, Cardiff RD, Carter JS, de Coronado S, Downing JR et al. Bethesda proposals for classification of nonlymphoid hematopoietic neoplasms in mice. Blood. 2002;100:238-45. PMID: 12070033 DOI: 10.1182/blood.v100.1.238. ↩
- Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, Long N, Schultz AR et al. Functional genomic landscape of acute myeloid leukaemia. Nature. 2018;562:526-531. PMID: 30333627 DOI: 10.1038/s41586-018-0623-z. ↩
- Faiola F, Liu X, Lo S, Pan S, Zhang K, Lymar E, Farina A, and Martinez E (2005). Dual regulation of c-Myc by p300 via acetylation-dependent control of Myc protein turnover and coactivation of Myc-induced transcription. Mol Cell Biol. 2005; 25:10220-10234. PMID: 16287840 DOI: 10.1128/MCB.25.23.10220-10234.2005. ↩