The tautomerization of porphycene on Cu(111) in simple physical terms
The tautomerization of porphycene on Cu(111) in simple physical terms
There are compounds, called isomers, that have the same molecular formulae but different molecular structures or different arrangements of atoms in space. In the so-called cis-trans isomerism, isomers have different positions of groups or specific atoms with respect to a double bond, a ring or a central atom.
For example, the numbers in the name 1,2-dichloroethene refer to the positions of the chlorine atoms; one chlorine atom is attached to carbon atom 1 and the other to carbon atom 2. Carbon 1 and carbon 2 are linked by a double bond. The structures of these isomers of 1,2-dichloroethene are
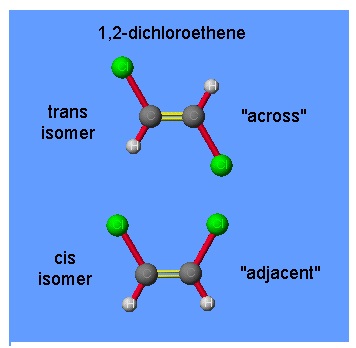
The carbon–carbon double bond is in reality one σ–bond and one π-bond. Because the characteristics of this bond it “locks” the two ends into a flat, rigid molecule, that can no rotate.
Cis and trans isomers have different properties. The cis isomer of 1,2-dichloroethene boils at 60 ºC, the trans compound at 48 ºC. These isomers can also be differentiated on the basis of dipole moment. The trans compound has no dipole moment because it is symmetrical (the polar C-Cl bonds point in opposite directions and so cancel). However, the cis compound definitely has a non-zero dipole moment.
If we wanted to transform one isomer into the other, what is called tautomerization, one end of the molecule must be rotated as the other remains fixed. For this to happen, the bond must be broken. Breaking the bond requires considerable energy, so the cis and trans compounds are not easily interconverted. Still, it is possible to convert one isomer to another if sufficient energy is supplied—say by chemical reaction. Or by electronic means.
Tautomerization reactions are particularly attractive because large changes in chemical reactivity are associated with simple intramolecular transformations. In this context, indirect adsorbate transformation by hot electrons has gained prominence as a tool to control the reactivity of species in the condensed phase.
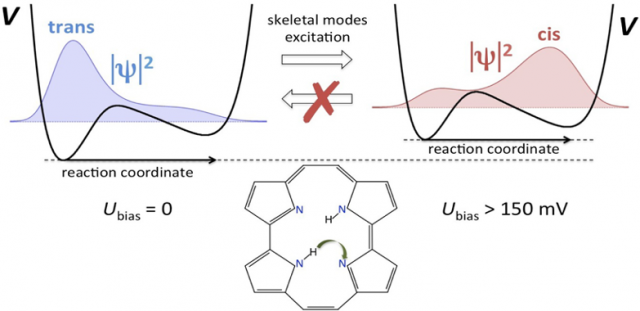
In recent years, much effort has been devoted to harness their potential as molecular switches, for which porphycene on Cu surfaces appears as a promising candidate. In particular, the trans → cis tautomerization of porphycene (a structural isomer of porphyrin) on Cu(111) was accomplished by injection of electrons via scanning tunnelling microscopy (STM). The process was observed only above a specific threshold voltage, below the resonant frequency of the N-H bond stretching mode, and in the vibrational band of the skeletal modes. The reaction takes place up to tens of nanometers away from the STM tip position, and the backward cis → trans tautomerization can be induced only by thermal activation. But the phenomenon is a mystery: no theory is able to explain all of these observations simultaneously.
Now, Dino Novko (DIPC), María Blanco-Rey (DIPC & UPV/EHU), and Jean Christophe Tremblay (FU-Berlin) propose 1 a microscopic dynamical model based on density functional theory (DFT) calculations that circumvents the need for high-dimensional quantum dynamics and rationalizes the experimental details of the STM-induced trans → cis tautomerization reaction in porphycene/Cu(111) in simple physical terms.

In the porphycene/Cu(111) system, the tautomerization reaction coordinate can be identified as H atom migration between the inequivalent imine and amine groups inside of the porphycene cavity (see Figure 2). The potential energy profile involves a low trans→cis activation barrier (∼150 meV) and a small energy difference (∼85 meV) between trans and cis. An important distortion of the molecular frame is observed along the tautomerization path leading to the cis configuration. A fully quantum dynamical description of a problem of such dimensions is beyond any conventional method.
According to the model, the STM creates hot electrons in the metal that are propagated as an S-wave through the substrate to reach the adsorbate. This accounts for the fact that tautomerization can be triggered in molecules far away from the injection point of the tunnelling electrons. The non-adiabatic coupling between hot surface electrons and the molecular vibrations indirectly distorts the potential non-uniformly along the reaction path via intermode coupling, thereby reversing the thermodynamical stability of the trans and cis configurations (see Figure 2). The H atom tunnels through the tautomerization barrier and relaxes rapidly through electron−hole pair coupling to the thermodynamically more stable cis configuration. At higher bias voltages, resonant excitation of the tautomerization mode increases the tunnelling rate along with the global reaction rate.
Thus, the model explains the experimentally observed stability reversal of the two configurations at a given threshold bias. The tautomerization rates are dominated by simultaneous tunnelling and relaxation to the cis ground state and dictated by the energetic alignment of the local vibrational ground states. The observed dependency of the tautomerization yield on the applied voltage can be rationalized from geometrical arguments based on the availability of electrons traveling as S-waves in the metallic substrate.
Because quasi-thermal fluctuations are suppressed at zero bias, the molecule remains trapped in the nearest local minimum of the potential energy landscape. The reverse cis → trans reaction can only be triggered by thermal activation, where it is mediated by hopping diffusion.
The model has a general character and could be applied to other similar systems that have already been investigated experimentally.
Author: César Tomé López is a science writer and the editor of Mapping Ignorance.
References
- Dino Novko, María Blanco-Rey, and Jean Christophe Tremblay (2017) Intermode Coupling Drives the Irreversible Tautomerization in Porphycene on Copper(111) Induced by Scanning Tunnelling Microscopy J. Phys. Chem. Lett. doi: 10.1021/acs.jpclett.7b00141 ↩