Starving the brain
Starving the brain
Life is sustained by chemical reactions, and the countless reactions that take place in the cells of living organisms that are responsible for every biological process are known as energetic metabolism. Among the others, the brain stands as the most energy-consuming organ, accounting for up to 20 % of the body’s total haul while representing only around 2 % of the total body weight 1. This implies that the brain continuously slurps up a huge amount of nutrients from the blood, due to its high energetic requirements. Metabolic activity is remarkably constant over time in the brain regardless of the “mindfulness” that your activity requires 2. To put it in easier words, your brain is craving for sugar all day long, whatever is that you are doing: sleeping, deeply meditating or solving an essential scientific problem.
How is all this energy obtained? Well, the preferred nutrient for the brain is the sugar glucose, a carbohydrate; therefore, neuronal activity relies largely on the transformation of glucose to carbon dioxide and water. This transformation results in the generation of great amounts of ATP: the body’s “energetic currency”. Energy in the form of ATP is spent in the brain to accomplish the different reactions that take place continuously. Due to the large energetic demand, the brain acts like a glucose “sink” receiving a vast blood supply.
However, the brain has one unusual property: it constitutes an extremely isolated environment. This is because it is separated from the rest of the body by a highly specialized tissue known as the meninges, and it is isolated from circulating blood thanks to the hematoencephalic barrier, aka blood-brain barrier (BBB). The BBB is a highly selective barrier that separates the blood from the brain extracellular fluid in the central nervous system. Hence, the BBB is important to protect the brain from infections, neurotoxins, and inflammation among other stressors. However, this barrier is disrupted in many disease conditions for example in stroke, traumatic brain injury and neurodegenerative diseases such as Alzheimer’s disease, multiple sclerosis, epilepsy or chronic HIV infection 3.
Therefore, the BBB is very important to protect the brain but complicates the delivery of nutrients to the brain. But how does glucose go across the barrier to feed the brain? There is a need of carriers to transfer nutrients and other molecules across the BBB. Among those, GLUT1 is a highly specialized glucose transporter, which is present in the endothelial cells that isolate the blood from the brain and allow the glucose to be transported and fuel brain activity.
As it seems, GLUT1 is very important, indeed, genetic defects in GLUT1 are the main cause of a pediatric disorder named De Vivo disease, aka GLUT1 deficiency syndrome (GLUT1 DS). GLUT1-DS is a rare condition inherited in a dominant fashion; this means that in GLUT1-DS patients one of the two copies of the GLUT1 gene is mutated. GLUT1 loss-of-function promotes an inadequate transportation of glucose across the BBB, which is as serious as it sounds: GLUT1-DS patients experience brain starvation. As a result, innumerable physical and mental disabilities take place commencing very early in the life of the child. The main characteristic is that these children have recurrent seizures. Also, they develop mental retardation, social and motor problems, ranging from mild to severe. In the worst cases, the child cannot walk nor talk 4. More than 500 patients with Glut1 mutations have been already described worldwide and there are reasons to believe that there are more cases going misdiagnosed or undiagnosed due to lack of awareness of the condition.
Is there any cure for this syndrome? As explained, GLUT1-DS results from the lack of glucose to adequately fuel brain activity; so, one could think that it could be solved by delivering a different nutrient to feed the brain. Bearing this in mind, a special diet has been used to treat the patients 5.
The ketogenic diet (KD) provides a much higher amount of fat than carbohydrates. The immediate effect is a change in the nutrient profile in the blood leading to the accumulation of ketone bodies. Ketone bodies constitute an alternative substrate for the brain that is directly metabolized thanks to its transport through a different kind of carriers present at the BBB, the monocarboxylate transporters, bypassing GLUT1 defect. Surprisingly, while KD eradicates seizures in patients, it does not ameliorate cognitive dysfunction, and mental retardation remains for patient’s life 6. Recent investigations have provided some light on the mechanisms that drive patient’s disability.
In order to model GLUT1-DS, the mouse gene that encodes the GLUT1 transporter, known as Glut1 has been deleted in a Glut1 “knock-out” (KO) model 7. A recent article 8 has connected the Glut1 defect to BBB disruption in Alzheimer’s disease (AD). For this investigation, both the Glut1 KO and an AD mouse model (APPSw/0) in which the toxic protein amyloid β was introduced, have been used. They found some similarities between the problems developed in GLUT1-DS and AD mice models. Unexpectedly, the reduced glucose transport into the brain in the Glut1 KO mice resulted in a problem in blood vessel development (angiogenesis) and BBB disruption at early stages. Later on, this mutant mice developed cognitive dysfunction and neurodegeneration. What happened when both defects were combined? In order to relate this to AD phenotype, the Glut1 deficient mice were bred with the APPSw/0 mice, and the result was that Glut1 deficiency worsened AD condition in the APP mice.
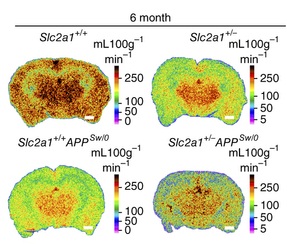
It was known that individuals at genetic risk for AD show a decreased glucose metabolism in the brain, even before showing any symptoms of the disease 9. Actually, the “brain starvation” that takes place at early stages of AD has been suggested to be one of the initial causes of the disease. However, the lack of experimental evidence left this as just a hypothesis until now. The novel findings about GLUT1 deficiency being especially deleterious in AD in mice models, together with new data in human showing that GLUT1 reduction takes place in AD patients (9) constitute a big step in the knowledge of the early mechanisms that drive Alzheimer’s neurodegeneration. GLUT1 constitutes a new therapeutic target for AD, a striking pathology in which huge research effort has not provided a treatment yet.
References
- Clark D. D. & Sokoloff, L. (1999) in Basic Neurochemistry: Molecular, Cellular and Medical Aspects, eds. Siegel, G. J., Agranoff, B. W., Albers, R. W., Fisher, S. K. & Uhler, M. D. (Lippincott, Philadelphia), pp. 637–670. ↩
- Sokoloff L., Mangold, R., Wechsler, R., Kennedy, C. & Kety, S. S. (1955) The effect of mental arithmetic on cerebral circulation and metabolism J. Clin. Invest.34,1101 ↩
- Gary A Rosenberg Neurological diseases in relation to the blood–brain barrier J. of Cereb. Blood Flow & Metab. (2012) 32, 1139–1151 ↩
- De Vivo, D. C., Trifiletti, R. R., Jacobson, R. I., Ronen, G. M., Behmand, R. A., Harik, S. I. Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. New Eng. J. Med. 325: 703-709, 1991. ↩
- Klepper, J., Scheffer, H., Leiendecker, B., Gertsen, E., Binder, S., Leferink, M., Hertzberg, C., Nake, A., Voit, T., Willemsen, M. A. Seizure control and acceptance of the ketogenic diet in GLUT1 deficiency syndrome: a 2- to 5-year follow-up of 15 children enrolled prospectively. Neuropediatrics 36: 302-308, 2005 ↩
- Gene Review: Glucose Transporter Type 1 Deficiency Syndrome http://www.ncbi.nlm.nih.gov/books/NBK1430/ ↩
- Wang D, Pascual JM, Yang H, Engelstad K, Mao X, Cheng J, Yoo J, Noebels JL and De Vivo DC A mouse model for Glut-1 haploinsufficiency Human Mol. Genet. Hum. Mol. Genet. 2006 15 (7): 1169-1179. ↩
- Winkler EA, Nishida Y, Sagare AP, Rege SV, Bell RD, Perlmutter D, Sengillo JD, Hillman S, Kong P,Nelson AR, Sullivan JS, Zhao Z, Meiselman HJ, Wenby RB, Soto J, Abel ED, Makshanoff J, Zuniga E, De Vivo DC & Zlokovic BV Nat. Neurosci. 18, 521–530 (2015). ↩
- Nordberg, A., Rinne, J.O., Kadir, A. & Långström, B. The use of PET in Alzheimer disease Nat. Rev. Neurol. 6, 78–87 (2010). ↩