A working molecular electronic switch
A working molecular electronic switch
A visionary perspective of molecular electronics is the use of single molecules as functional entities in electronic devices. In this context, some of the most important components are molecular switches, which are molecules that can be interconverted reversibly between at least two stable states, one for the OFF and one for the ON.
In order to characterize and manipulate single atoms and molecules on surfaces scanning tunneling microscopy and spectroscopy (STM/STS) represents an ideal tool. Using it several switching mechanisms have been explored previously with molecular and submolecular resolution and have been induced locally by different means: tunneling electrons, applied electric fields, or by combined stimuli in the STM tunnel junction. The changes of the molecular configurations that these switching mechanisms provoke can be quite diverse, from the folding in complex organic molecules, and different types of isomerization processes to changes of the atomic position in metalorganic complexes, and the formation and dissociation of chemical bonds. Any of these conformational changes may be accompanied by charging. and discharging processes.
Recently, another scenario was proposed theoretically in which a switching process of an individual molecule is related to bistable electronic states. These are associated with different bonding strengths and hybridization degrees of the molecules to a metallic substrate, resulting in a change of the bending of the molecular conformation associated with a change of the adsorption distance.
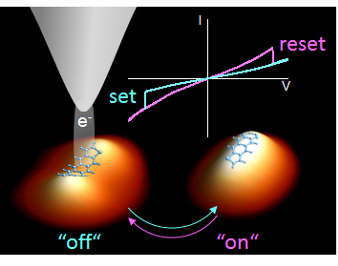
Now, a group of researchers that includes Rémi Pétuya (DIPC) and Andrés Arnau (DIPC and UPV/EHU) has found1 single molecular switches consisting of individual anthradithiophene (ADT) molecules adsorbed on Cu (111). ADT is a structural analogue of pentacene, with two sulfur-containing groups at each molecular terminal side representing cis- and trans-diastereomers.The switching of individual molecules is activated locally by injecting charge carriers from the STM tip apex into the molecule.

Their first-principles calculations using van der Waals density functionals where exchange and correlation are treated consistently allow them to propose a model based on the appearance of two adsorption states corresponding to two different geometric (bent and planar) and electronic conformations. These two conformations are induced by two main driving forces, the S-Cu bond length and the adsorption constraint for an optimal binding of the acene groups to the substrate, similar to pentacene.
The molecules are reversibly interconverted by voltage pulses of opposite polarity between two different adsorption geometries that correspond to two different electronic conformations of the molecule. The characteristic parameter responsible for the structural and electronic changes is the adsorption distance that determines the bonding strength and the degree of hybridization between the ADT molecule and the surface.
The most stable geometry, the OFF state, which all molecules adopt upon evaporation, corresponds to a short adsorption distance at which the electronic interactions of the acene rings bend the central part of the molecule toward the surface accompanied by a significant charge transfer from the metallic surface to the ADT molecules. In the ON state the molecule has a flat geometry at a larger distance from the surface; because of this the interaction is weaker, resulting in a negligible charge transfer with an orbital structure resembling the highest occupied molecular orbital. The potential barrier between these two states can be overcome reversibly by injecting charge carriers locally into individual molecules.
These findings may be transferred to potential storage bits, allowing for a controlled switching to ON and OFF states at the individual molecular level.
Author: César Tomé López is a science writer and the editor of Mapping Ignorance.
References
- Bogdana Borca, Verena Schendel, Rémi Pétuya, Ivan Pentegov, Tomasz Michnowicz, Ulrike Kraft, Hagen Klauk, Andrés Arnau, Peter Wahl, Uta Schlickum, and Klaus Kern (2015) Bipolar Conductance Switching of Single Anthradithiophene Molecules ACS Nano DOI: 10.1021/acsnano.5b06000 ↩