The recycling of data unveils genomic regions related to celiac disease
The recycling of data unveils genomic regions related to celiac disease
Author: Koldo Garcia-Etxebarria got his Ph.D. in genetics at UPV/EHU in 2010. After a couple of years at the Evolutionary Biology Institute (CSIC-UPF) in Barcelona, he returned to UPV/EHU and joined José-Ramón Bilbao’s lab where he researches celiac disease.
Celiac Disease (CD) is an autoimmune disease that is developed in susceptible individuals when the gluten is present in their diet. As consequence, there is an inflammation of the small intestine, what has different effects, such as pain and diarrhoea, and the only effective treatment known is a gluten-free diet. A great portion of the susceptibility to CD is genetic. The major contributors are the DQ genes, located in the Major Histocompatibility Complex, a genomic region related to immune response. However, although these genes are essential to develop CD, are not enough to explain the whole genetic component of CD 1.
A way to find the missing genetic component of complex traits or diseases such as CD, are the Genome-Wide Association Studies (GWAS). In these studies, thousands of genetic markers (called single nucleotide polymorphisms, SNPs) across genome are analysed and their frequency is calculated in individuals with the trait of interest and individuals without that trait to find if any of these markers is more frequent (or scarcer) in one or another population. In this way, new variants related to the trait of interest could be detected without previous assumptions.
In the case of CD, different efforts based on association analyses have been carried out to unveil the rest of genetic component [1]. Among them, an association study using the so-called Immunochip platform, a genotyping array which is enriched with SNPs from genomic regions related to immune-mediated diseases. In the case of CD 12041 celiac and 12228 healthy controls where analysed and 40 genomic regions were linked to CD. In that study individuals from 6 countries (United Kingdom, Spain, Netherlands, Italy, Poland and India) were studied and the heterogeneity due to the different origin of the individuals was corrected using the geographical origin as variable. 2
However, when our group analysed the individuals which were provided to that study (545 celiac and 308 controls) some discrepancies arose. Thus, we ask ourselves whether the different origin of the individuals and their different proportion (the 66% of individuals were from UK) were causing a bias. To overcame this bias that could be present in the original study due to the great heterogeneity of the analysed samples, our group analysed the same data used in the original Immunochip paper making some slight corrections and using an innovative approach 3 after asking permission to the Welcome Trust Case Control Consortium.
First, we defined conserved blocks of SNPs to avoid the variability that recombination could cause and one SNP from each block randomly was selected. These SNPs were then used to calculate the ancestry of the individuals. This is a technique used in population genetics to calculate the mixture of individuals from a group or population and we thought that could be useful to cluster individuals depending on their genetic ancestry rather than geographical origin. Our analysis revealed that the individuals could be clustered in 30 groups.
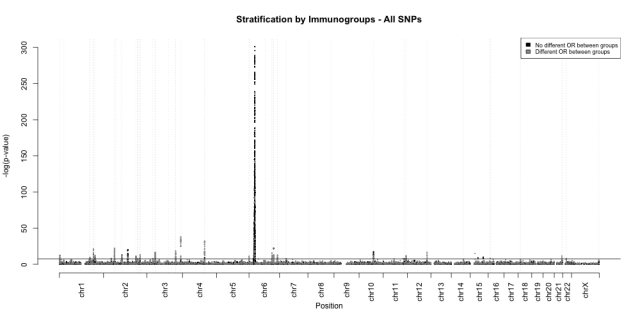
Then, we carried out an association analysis with all SNPs analysed in the Immunochip, using the Cochran-Mantel-Haenszel test, a statistical test that takes into account the stratification in different groups. We compared our results with the original ones of the Immunochip and we detected a new genomic region related to CD and extended other known regions (Figure 1). However, we were not able to detect two genes associated in the original analysis of the Immunochip, although other authors have stated their concerns about those genes.
With the aim to validate our results, and to prove the suitability of our approach based on genetic ancestry, we further analysed the genes in the new region and the ones located in the extended regions. For that, in collaboration with the Paediatric Unit of Cruces University Hospital and with all the bioethical approvals, we analysed the expression of the genes in biopsies from the duodenum, the tissue where the disease is developed, of patients with CD, these patients after two years of gluten-free treatment and healthy controls. We compared the expression levels of 14 genes (11 located in the new region, 2 from extended regions and 1 located in a solitary signal) in the different conditions and we found significant differences in 9 of them in any of the comparisons. In fact, two genes from the new region did not recover their expression level after gluten-free treatment, pointing out a malfunction of these genes in susceptible individuals. In addition, the lactase gene was located in these new region. It was previously known that patients with CD were not able to take milk and, therefore, we found the link between genomic variants and the less expression of lactase. Since its expression was recovered after gluten-free treatment, the lower expression is a consequence of the disease. Thus, our approach reanalysing existing data was successful and we were able to find new candidate genes that were truly related to the disease.
In the future, we expect that this approach could be use in other complex diseases. Data of different diseases is available for the researchers and, since we were able to find new candidate genes, it seems worth the reanalysis of other diseases using new approaches, such as based on genetic ancestry. In this way, genes related to diseases that are hidden could arise and we could better understand the genetic basis of complex diseases.
References
- Ricaño-Ponce, I. et al. (2015) Genetics of celiac disease. Best Pract Res Clin Gastroenterol 29, 399–412, doi:10.1016/j.bpg.2015.04.004 ↩
- Trynka, G. et al. (2011) Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat Genet 43, 1193-1201, doi:10.1038/ng.998 ↩
- Garcia-Etxebarria, K. et al. (2016) Ancestry-based stratified analysis of Immunochip data identifies novel associations with celiac disease. Eur J Hum Genet in press, doi:10.1038/ejhg.2016.120 ↩