Desorption as a first-order phase transition
Desorption as a first-order phase transition
Adsorption commonly is understood as the reversible binding of molecules and atoms from the gaseous or liquid phase on surfaces, mostly of highly porous adsorbent media. In chemisorption a single layer of molecules, atoms or ions is attached to the adsorbent surface by chemical bonds; in physisorption only van der Waals forces are involved. Adsorption is a ubiquitous phenomenon affecting the properties of all types of interfaces.
The technical applications of adsorption cover a wide range, such as separation, synthesis, storage, purification of gases and liquids, and even air conditioning. It is increasingly becoming established in chemical processes as an alternative to conventional separation techniques, and in catalysis. It is apparent that all these technical applications are considerably based on a better understanding of the adsorption mechanisms.
While approaching an adsorbing wall, particles lose their translational and rotational freedom and rearrange by adopting spatial configurations differing from those far from the interface. The related changes in particle density at the boundary between adsorbent and adsorbate determine the performance of the whole adsorbed layer.
In the last decade, some striking correlations identified in adsorbed polymer layers between the number of chains adsorbed and a variety of properties have been found. Glass transition temperature, viscosity, mechanical properties, lateral diffusion, crystallization rate, and maximum water uptake, are just a few examples of the variables affected by the number of adsorbed chains. If we could control this number in a simple and reliable industrial way, like using simple thermal treatments, we could design new processing strategies to finely tune the properties of nanomaterials. We need to know more about the thermodynamics and the mechanisms of adsorption and desorption in order to achieve this control
Polymers adsorb more easily than small molecules. Actually, while adsorption is still reversible at the monomer level, desorption of a whole chain is less likely to occur, because it requires simultaneous detachment of numerous adsorbed monomers. As the probability that such an event would occur within the timescales and at the temperatures of technological interest is very low—that is, the energetic barrier for desorption is very large—, for practical purposes, chain adsorption is considered as irreversible.
This reasoning is, however, merely based on kinetics as is easy to see. The adsorbed amount (or equivalently the thickness of an adsorbed layer) is, in fact, an equilibrium quantity related to the amount of free chains present in the environment, e.g., the concentration of a polymer solution. The number of adsorbed chains increases with polymer concentration until reaching a saturation value, given by interfacial interactions. Desorption of adsorbed chains is, hence, possible by placing a polymer layer in a pure solvent, or in a solution whose concentration is lower than that related to the equilibrium adsorbed amount. Importantly, the concentration thresholds to stabilize adsorbed layers are rather low, as corresponds to weak desorption forces and extremely long desorption times: soaking for 4 months in pure chloroform is not sufficient to fully desorb an adsorbed layer of polystyrene (PS).
From a thermodynamic point of view, this phenomenon can be depicted as a phase transition of a non-adsorbed “bulk” polymer melt into a tightly adsorbed layer. This transformation entails a reduction of enthalpy and, to a smaller extent, also of entropy. As a consequence, with increasing temperature the larger weight of entropic effects is expected to equalize the free energy of the two phases, thereby reverting the transformation and the opposite phenomenon, that is, desorption of polymer chains from the solid substrate, should be observed. At the temperature at which desorption occurs, a phase transition is, therefore, expected.
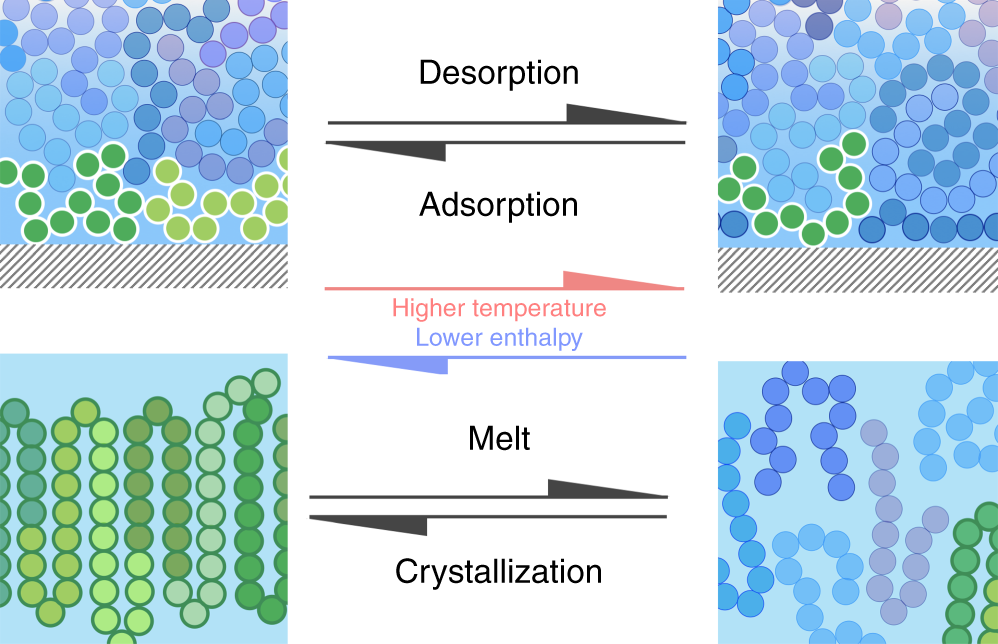
Even though desorption is a phenomenon as relevant as adsorption, a signature of desorption of a polymer melt from a solid substrate has so far remained unidentified. The main reason is that polymers heated at standard rates (~1–10 K min−1) generally undergo degradation before desorption.
To overcome this problem, a team of researchers has now employed 1 fast scanning calorimetry, which can be used to determine the calorimetric response of thin polymer layers processed at 104 K s−1. Applying such rate to adsorbed polymer layers permits avoiding degradation and, therefore, clear-cut evidence of desorption of a polymer melt is obtained.
This way the researchers demonstrate two things. First, that it is possible to investigate, using fast scanning calorimetry, the desorption of a polymer melt from a solid substrate. The experiments were performed on thin layers of poly(4-tert-butylstyrene) and polystyrene previously adsorbed on silicon oxide.
And second, that most of the enthalpy of desorption is related to the conformational changes occurring at the liquid/liquid phase transition. The enthalpy and temperature of desorption are independent of the annealing temperature, which indicates that adsorption / desorption is a first order thermodynamic transition. In other words, desorption can be treated as a first-order phase transition, in ways analogous to melting.
Author: César Tomé López is a science writer and the editor of Mapping Ignorance.
Disclaimer: Parts of this article might have been copied verbatim or almost verbatim from the referenced research paper.
References
- Monnier, X., Napolitano, S. & Cangialosi, D. (2020) Direct observation of desorption of a melt of long polymer chains. Nat Commun doi: 10.1038/s41467-020-18216-y ↩