Controlling the magnetic anisotropy of a transition metal complex
Controlling the magnetic anisotropy of a transition metal complex
As candidates for high-density data storage and quantum computation, magnetic transition metal complexes have attracted extensive attention. It has been established that the atomic surroundings of the magnetic ions play a significant role and govern the magnetism of these magnetic complexes. One feature in particular, magnetic anisotropy, which describes the preferential orientation of the magnetic moment in certain directions, arises from the crystal field and the ligand field, which make the degenerate d-orbitals of the transition-metal ion (they have equal energy, but different spatial arrangements, when the ion is isolated) to have different energies via spin−orbit coupling. Understanding and controlling the magnetic anisotropy of the magnetic complexes are most important for practical applications.
Till now, a few approaches to tune the magnetic anisotropy of adsorbed coordination complexes have been reported, such as altering the ligand field via chemical modification or mechanical deformation. Chemical modification often introduces irreversible changes to the metal complexes and prevents a reversible manipulation of the magnetic anisotropy. A mechanical deformation, on the other hand, remains in place only as long as the external force used to produce it is maintained and controlling numerous molecules is difficult.
As these complexes are usually adsorbates, could a manipulation of the adsorption process alter magnetic anisotropy? The variation of the molecular adsorption site does not suffer from the limitations from chemical modification or mechanical deformation, but so far little is known about the site dependence of the magnetic anisotropy energy of molecules.
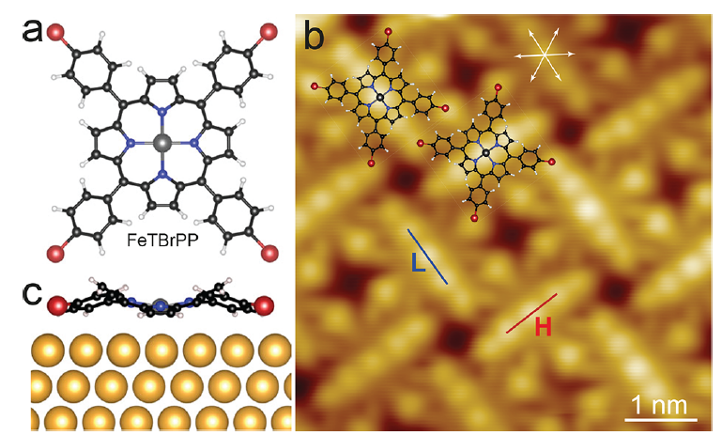
Now, a team of researchers has explored 1 the behaviour of the magnetic complex Fe(II) 5,10,15,20-tetrakis(4′-bromophenyl)porphyrin (FeTBrPP) when adsorbed on Au(111) with a low-temperature scanning tunnelling microscope (STM) and density functional theory (DFT). The molecule has two stable adsorption structures on the surface that differ in magnetic anisotropy energy. The structural difference also leads to a distinct peak shift of the molecular orbitals.
FeTBrPP molecules exhibit two orientations, either parallel (L) or perpendicular (H) to a close-packed direction of a Au(111) substrate and occupy top and bridge sites, respectively. Different electronic structures and magnetic anisotropy energies are observed for the two adsorption geometries. DFT calculations reveal that the structural differences affect the distance between the Fe ion and substrate atoms and lead to variations of the Fe 3d level occupations, which in turn change the axial magnetic anisotropy.
Importantly, the researchers also show that the magnetic anisotropy energy of the molecules can be easily tuned by manipulating the adsorption structure or the distance between the STM tip and the molecule. Actually, the contact of the STM tip to the molecular centre leads to an abrupt drop of
the anisotropy. This way, the magnetic anisotropy of the molecules can be varied by switching between H and L adsorption geometries that are equally stable on the surface.
The fact that the magnetic anisotropy energy can be controlled by changing the adsorption site, the orientation, or the tip−molecule distance is the kind of things that are found useful for spintronic applications.
More on the subject:
Open-shell organic systems that contain magnetically active transition metal ions
Modified adsorption geometry preserves the topological surface state
Controlled molecular rotors mounted on a molecular platform on a gold substrate
Author: César Tomé López is a science writer and the editor of Mapping Ignorance
Disclaimer: Parts of this article may have been copied verbatim or almost verbatim from the referenced research paper/s.
References
- Xiangzhi Meng, Jenny Möller, Masoud Mansouri, Daniel Sánchez-Portal, Aran Garcia-Lekue, Alexander Weismann, Chao Li, Rainer Herges, and Richard Berndt (2022) Controlling the Spin States of FeTBrPP on Au(111) ACS Nano doi: 10.1021/acsnano.2c09310 ↩