PHF6 maintains acute myeloid leukemia via regulating NF-κB signaling pathway
PHF6 maintains acute myeloid leukemia via regulating NF-κB signaling pathway
Author: Marta Irigoyen is a postdoctoral researcher at CIC bioGUNE
Acute myeloid leukemia (AML) is a major hematopoietic malignancy characterized by the uncontrolled expansion of the immature myeloid cells 1. The 5-year survival rates of young patients are 40–50% and less than 15% for patients aged over 60 2. For this reason, a great effort has been made to decipher the molecular events underlying AML transformation with the goals to identify specific therapeutic targets and develop new and more effective drugs. In this regard, since the epigenetic dysregulation is a recurrent event in leukemogenesis 3, it would stimulate oncogenic transcriptional programming that are important for AML initiation and progression.
Plant homeodomain finger gene 6 (PHF6), is a highly conserved epigenetic transcriptional regulator that plays critical roles in neurodevelopment and hematopoiesis. Interestingly, PHF6 mutations occur in 3% of acute myeloid leukemia 4 but the exactly functional role of PHF6 in human AML remain unknown. In the current study, Hou S. and collaborators found that PHF6 is essential for the maintenance of self-renewal ability of leukemia stem cells (LSCs) but dispensable for hematopoietic stem cells (HSCs) in vivo. Besides, the survival of leukemia cells is sensitive to PHF6 depletion, suggesting that PHF6 may be a potential LSC-directed therapeutic target for AML 5.
Firstly, to investigate the potential role of PHF6 in AML, authors analyzed the relationship of PHF6 expression and overall survival and showed that AML patients with high PHF6 expression had unfavorable prognosis than AML patients with low PHF6 expression. To further confirm whether PHF6 was truly functional relevant to myeloid leukemia development, they over-expressed PHF6 in AML cell lines and found that overexpression of PHF6 increased the growth and decreased the apoptosis of AML cells. Corroborating these data, they showed that PHF6 inhibition significantly decreased cell growth, promoted cell apoptosis and decreased the clonogenic potential of AML cells. Furthermore, the silencing of PHF6 in CD34+ cord blood cells had no influence on hematopoiesis. Thus, these results suggested that PHF6 was essential for the proliferation of myeloid leukemia cells, but dispensable for normal hematopoiesis.
Then, to determine the impact of PHF6 deletion in leukemogenesis in vivo, they utilized two mouse models showing that PHF6 abolition was able to decrease the number of leukemic cells in both peripheral blood and bone marrow. Furthermore, PHF6 deletion decreased extramedullary cell infiltration, increased the animal lifespan (survival time) and promoted the differentiation of leukemia cells. Taken together, these results demonstrated that PHF6 deficiency delayed AML progression in vivo.
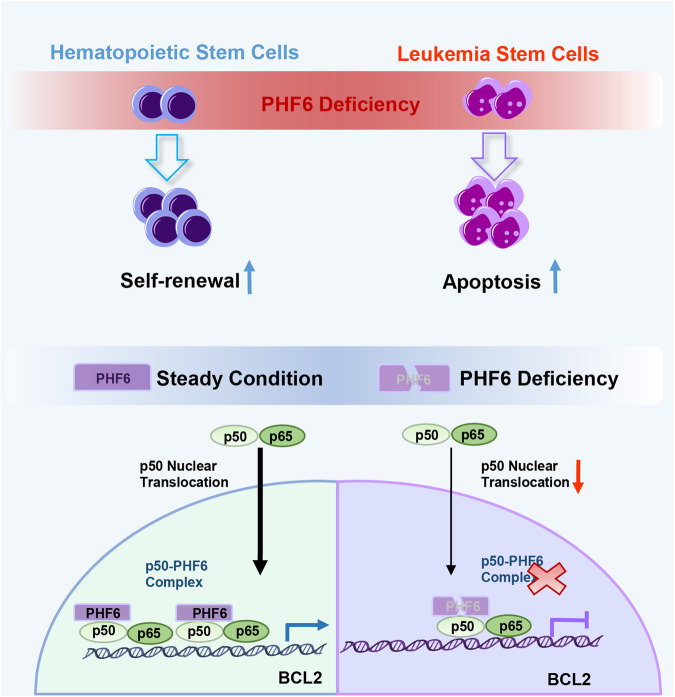
To determine the consequence of PHF6 loss of the stem cell subpopulation of leukemia cells (abbreviated as “LSCs” from “Leukemia Stem Cells”), and particularly on maintenance of in AML in vivo, they characterized LCSc in two AML mouse models. They used them in an advanced stage of the disease when the AML mice became moribund. Interestingly, they found that the number of LSCs were decreased in bone marrow (BM), spleen and liver of PHF6-deficient mice as compared with that of wild type (WT) mice. To quantify the functional LSCs in vivo, they performed extreme limiting dilution transplantation assays with AML cells from PHF6-deficient or WT mice, and observed that the frequency of LSCs was reduced in recipient mice grafted with PHF6-deficient AML cells. In summary, these data indicated that loss of PHF-6 effectively delayed the AML progression by impairing the self-renewal capacity of LSCs.
The underlying molecular mechanisms of PHF-6 loss
To explore the underlying molecular mechanisms of PHF-6 loss in delaying AML progression, the authors analyzed the transcriptional profiles of PHF6-deficient AML cells respect normal AML cells via RNA sequencing. This comparison revealed a distinct gene expression signature in PHF6-deficient cells. Gene analysis showed that leukemogenesis-related pathways were downregulated, myeloid cells development and homeostasis-related pathways were upregulated, and apoptosis and myeloid cells development-related genes were upregulated. Interestingly, TNFα-NF-κB and chemokine-regulated genes were downregulated in PHF6-deficient cells. Notably, PHF6 loss inhibited the NF-κB signaling pathway in murine and human AML cells when compared with WT AML cells. Thus, these results suggested that PHF6 depletion might promote AML cell apoptosis by blocking the NF-κB signaling pathway.
To investigate the intracellular mechanisms by which PHF6 regulated NF-κB activity in human AML, the authors assessed the expression of NF-κB related factors in PHF6-deficient human AML cells and control cells. Thus, they found that the expression of p-IKKβ/IKKβ (a kinase complex that activates NF-κB signaling), p50 and p-p65/p65 (the most abundant form of NF-κB is the heterodimer formed by the p50 and p65 subunits) were not altered in PHF6-deficient human AML cells when compared with their respective controls. Since the nuclear translocation of p50 and p65 directly regulates NF-κB downstream target genes, they further assessed the nuclear translocation of p50 and p65 in PHF6-silenced myeloid leukemia cells in response to NF-κB activation through TNFα treatment. They found that in control cells p50 was able to successfully translocate into the cell nucleus, however, in PHF6-deficient human AML cells was retained at the cytoplasm. Furthermore, they found that PHF6 overexpression led to more p50 translocated from the cytoplasm to the nucleus under the stimulation of TNFα. In this process, PHF6 accompanied p50 into the nucleus and eventually co-localizing with it.
A physical interaction
To further investigate whether PHF-6 directly bound with p50 and regulated the function of p50, they modified genetically cells in which PHF6 and p50 were marked and overexpressed. Thus, performing co-immunoprecipitation experiments (a technique to isolate protein complexes) they confirmed a physical interaction of PHF6 and p50. Furthermore, it has been reported that p50 directly binds to BCL2 gene (which encodes a protein that blocks apoptosis) promoting its transcription. They found that the mRNA expression of BCL2 was decreased in PHF6-deficient cells and was increased in PHF6 overexpressing ones when compared with WT condition. These data demonstrated that PHF6 deficiency blocked NF-κB signaling pathway by disrupting the PHF6-p50 transcriptional complex and partially inhibiting the p50 nuclear translocation.
Finally, to determine whether inhibition of NF-κB could delay the overproliferation of PHF6 overexpressing cells, they treated PHF6 overexpressing myeloid leukemia cells with BAY11 7082 (a selective NF-κB inhibitor) and found that BAY11-7082 effectively reduced the mRNA expression of BCL2 and BCL-XL (anti-apoptotic genes). These results indicated that this drug was able to inhibit succesfully the activity of NF-κB signaling pathway. Besides, they found that BAY11-7082 significantly induced the apoptosis and inhibited the growth of PHF6 overexpressing cells in vitro. Next, they assessed the anti-leukemia efficacy of the NF-κB inhibitor on PHF6 overexpressing cells in vivo. They transplanted PHF6 overexpressing AML or control cells into NSG mice and treated these mice with BAY11-7082. Upon treatment with BAY11-7082, the mice engrafted with PHF6 overexpressing cells showed slight infiltration of AML cells in the BM than that of mice engrafted with control cells, indicating that BAY11-7082 could partially rescue the over-proliferation of PHF6 overexpressing cells in vivo. In addition, they found that apoptotic cells were increased in BM of mice engrafted with PHF6 overexpressing cells than that of mice engrafted with control cells. These results demonstrated that a potential therapy using NF-κB inhibitors could reduce the progression of PHF6 overexpressing AML cells by promoting leukemia cells apoptosis in vivo.
In summary, the authors demonstrated that: 1) PHF6 maintains AML via regulating NF-κB signaling pathway and 2) a treatment of PHF6 over-expressed myeloid leukemia cells with a NF-κB inhibitor significantly increased tumor cell apoptosis and decreased their proliferation. Taken together, they found that PHF6 plays a pro-oncogenic role in myeloid leukemia and could be a potential therapeutic target for treating myeloid leukemia patients.
References
- Estey E, Döhner H. Acute myeloid leukaemia. Lancet 2006; 368: 1894-907. PMID: 17126723 DOI: 10.1016/S0140-6736(06)69780-8. ↩
- Kantarjian HM, Kadia TM, DiNardo CD, Welch MA, Ravandi F. Acute myeloid leukemia: Treatment and research outlook for 2021 and the MD Anderson approach. Cancer 2021; 127: 1186-1207. PMID: 3373444 DOI: 10.1002/cncr.33477. Epub 2021 Mar 18. PMID: 3373444. ↩
- Rodríguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med 2011;17:330-9. PMID: 21386836 DOI: 10.1038/nm.2305. ↩
- Van Vlierberghe P, Patel J, Abdel-Wahab O, Lobry C, Hedvat CV, Balbin M, Nicolas C, Payer AR et al. PHF6 mutations in adult acute myeloid leukemia. Leukemia. 2011 Jan;25(1):130-4. PMID: 21030981 DOI: 10.1038/leu.2010.247. ↩
- Hou S, Wang X, Guo T, Lan Y, Yuan S, Yang S, Zhao F, Fang A et al. PHF6 maintains acute myeloid leukemia via regulating NF-κB signaling pathway. Leukemia 2023; 37: 1626-1637. PMID: 37393343 DOI: 10.1038/s41375-023-01953-6. ↩