Targeting mitochondrial structure sensitizes acute myeloid leukemia to Venetoclax treatment
Targeting mitochondrial structure sensitizes acute myeloid leukemia to Venetoclax treatment
Author: Marta Irigoyen is a postdoctoral researcher at CIC bioGUNE
Acute Myeloid Leukemia (AML) is a hematopoietic neoplasm characterized by the proliferation and accumulation of aberrant immature myeloid progenitor cells. AML is associated with poor clinical outcome and high mortality, with an overall five-year survival rate of less than 15-30%. For AML patients, standard therapies often fail to achieve the complete remission, disease relapse is often fatal and bone marrow transplantation are only applicable to a few selected AML patients, highlighting the need for novel targeted treatments. A number of such emerging treatments have targeted essential “hallmarks” of cancer, including the regulation of cancer cell survival by members of the BCL-2 protein family. Among these members, B-cell lymphoma 2 (BCL-2) has been found to be upregulated in AML cells 1 and, specifically, in leukemic stem cells (LSC) 2. Besides, BCL-2 overexpression is a poor-prognosis factor in AML and is associated with poor response to standard cytotoxic therapy 3.
Mechanistically, BCL-2 prevents programmed cell death by binding to pro-apoptotic members of the same family 4. In cancer cells, enhanced BCL-2 expression supports cell survival, as it leads to the suppression of mitochondrial-mediated apoptosis. Inhibiting BCL-2 via a selective BH3 mimetic (a domain necessary for the pro-apoptotic function) such as Venetoclax has proven to be an efficient strategy to promote caspase-dependent cell death in AML 5. However, approximately 30% of patients do not respond to treatment and many AML patients develop resistance 6. Thus, to shed light on the mechanisms of resistance to Venetoclax, Xufeng Chen and coworkers identified genes whose inactivation sensitizes AML blasts to improve this new drug treatment 7.
They firstly carried out a genome-wide CRISPR/Cas9 screening (a tool that enables genome-wide interrogation of gene function) 8 in human AML cells in the presence or absence of Venetoclax. The results revealed potential modes of resistance to Venetoclax drug. They were particularly interested in the negatively selected genes, as they were potential therapeutic targets for circumventing Venetoclax resistance. Thus, gene analysis of the “sensitizer” genes revealed strong enrichment of genes that encode proteins functioning in mitochondria (a cellular organelle critical for energy generation), highlighting the relevance of mitochondrial processes in the sensitization of AML cells to Venetoclax.
Given that Venetoclax action converges on mitochondria which actively participate in programmed cell death, they aimed to further investigate how BCL-2 inhibition impacts mitochondrial structure and function in AML. Mitochondria change their shape early during the process of apoptosis and indeed, mitochondria exhibit abnormal ultrastructural morphology after Venetoclax treatment. Since at the beginning of the apoptosis (cell suicide), the mitochondrial membrane potential collapses leading to the complete cytochrome c release from mitochondria to the cytosol (9), authors predicted that Venetoclax (as an inducer of apoptosis), will cause the loss of mitochondrial membrane potential in leukemic cells. Then, they focused on the mitochondrial protein called “Optic Atrophy 1” (OPA1), which functions as a molecular staple at cristae junctions (regions of the mitochondria that works as diffusion barriers for Ca2+ and H+ ions) to prevent cytochrome c mobilization and release 9. Interestingly, upon an intrinsic death stimulus, OPA1 proteolytical cleavage is enhanced allowing the “opening” of the cristae junctions and the cytochrome c redistribution 10.
To further investigate the mechanisms of Venetoclax resistance, authors generated human AML clones highly resistant to the drug. Since MCL-1, an anti-apoptotic member of the BCL-2 family, upregulation has been previously implicated in the development of Venetoclax-resistance 11, they aimed to examine the protein levels of the major BCL-2 family members (MCL-1, BCL-2 and BCL-XL) in the generated Venetoclax-resistant cell lines. Although they observed a slight increase of MCL-1 and BCL-2 protein levels in these cells, OPA1 upregulation and mitochondrial structural adaptations appear to represent a more universal mechanism of Venetoclax resistance. To further explore the molecular mechanisms underpinning Venetoclax resistance, they performed RNA-sequencing and, among the differentially expressed genes, they focused on the genes involved in mitochondrial processes. Strikingly, pathways that regulate mitochondrial membrane organization, potential and depolarization were strongly enriched in Venetoclax-resistant cells. Besides, metabolic processes of key cellular components were also enriched, suggesting the metabolic adaptation is likewise important for Venetoclax resistance.
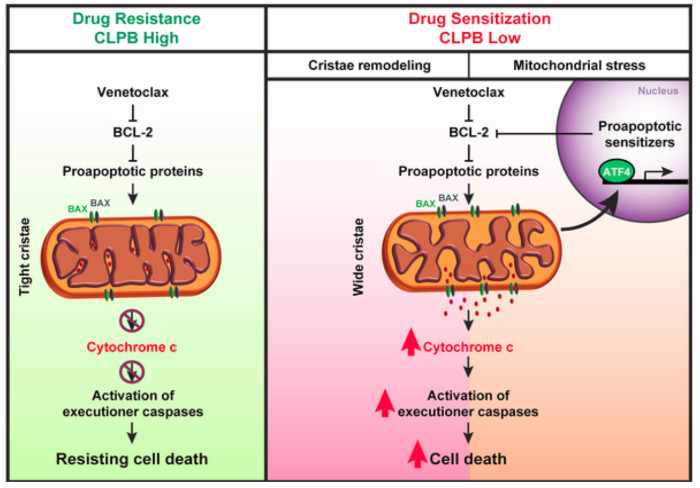
Next, they attempted to determine whether there is a correlation between genes differentially expressed in the resistant cell lines and genes-hits in their CRISPR screen. Since they identified genes encoding different mitochondrial proteins, they hypothesized that targeting these proteins might open up potential synthetic lethal vulnerabilities for Venetoclax treatment. Refining the candidate list from the screen, they focused on genes that displayed higher mRNA expression levels in AML patient samples compared to healthy primary CD34+ hematopoietic stem and progenitor cells (HSPCs). After applying these criteria, one of the top-scoring candidates was CLPB, that encodes a mitochondrial chaperonin that is also overexpressed in Venetoclax-resistant human AML cell lines. To further evaluate the importance of CLPB function, they depleted CLPB in AML cell lines and showed that CLPB depletion significantly reduced the IC50 of Venetoclax, confirming that CLPB ablation can sensitize AML cells to Venetoclax treatment. Finally, in vivo studies corroborated that CLPB deletion synergizes efficiently with Venetoclax in both Venetoclax-sensitive and resistant xenografts.
Then, they aimed to investigate how CLPB loss promotes Venetoclax-induced programmed cell death in AML cells. Since they observed that CLPB co-localizes with a mitochondrial marker in AML cells, they hypothesized that CLPB participates in the organization of mitochondrial ultrastructure. Interestingly, mitochondria lacking CLPB exhibited an aberrant structure which were exacerbated when cells were treated with Venetoclax. Besides, the structural perturbations of CLPB-ablated mitochondria were accompanied by a prominent accumulation of short OPA1 forms generated after OPA1 proteolytical cleavage, indicating excessive OPA1 processing. Next, they confirmed the physical interaction of CLPB with OPA1 suggesting that CLPB regulates mitochondrial cristae morphology and apoptosis in AML cells, via its specific interactions with OPA1. Finally, they examined the in vivo significance of CLPB in AML progression and observed a significant delay in leukemia progression in mice receiving CLPB-deficient AML cells.
To further focus on the underlying mechanisms of CLPB function, they performed RNA-Sequencing in AML cells upon CLPB deletion. Pathway analysis of the differentially expressed genes in CLPB-deficient AML cells showed strong enrichment on metabolic pathways. Data analysis to reveal the upstream transcriptional regulators that are responsible for the transcriptional landscape changes upon CLPB ablation showed that ATF4 12 ranked first in their analysis. Given that ATF4 has been identified as a key regulator of the mitochondrial stress response in mammals 13, they speculated that CLPB-deficient AML cells are under mitochondrial stress. Gene-Set Enrichment Analysis (GSEA) revealed that genes associated with mitochondrial stress response were indeed strongly enriched in CLPB-deficient AML cells. To functionally confirm the mitochondrial stress in CLPB-deficient AML cells, they measured mitochondrial reactive oxygen species (ROS) after the treatment with a complex III inhibitor and the results indicated greater ROS accumulation inside CLPB ablated mitochondria upon challenge. Considering that CLPB-deficient AML cells are under mitochondrial stress, they did a more detailed metabolic profiling of these cells that revealed a significant enrichment in amino acid related pathways.
Finally, since resistance to Venetoclax monotherapy rapidly ensues in AML 1415, multiple combinational therapies for Venetoclax have been proposed and tested. Among these, the combination of Venetoclax with hypomethylating agents (HMA), like azacitidine, is currently in clinics due to its favorable responses in the clinical trials 16. Thus, they proposed to investigate if CLPB inhibition can enhance the efficacy of Venetoclax and Azacitidine combined treatment, and they confirmed that CLPB ablation sensitized AML cells to the combined treatment.
In conclusion, their findings highlight that loss of the mitochondrial protein CLPB leads to structural and functional defects of mitochondria, hence sensitizing AML cells to apoptosis. Therefore, targeting CLPB synergizes with Venetoclax and Venetoclax/Azacitidine combination in AML and may be a future novel therapeutic approach to treat the fatal disease.
References
- Campos L, Rouault JP, Sabido O, Oriol P, Roubi N, Vasselon C, et al. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood. 1993;81:3091-6. PMID: 7684624. ↩
- Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12:329-41.PMID: 23333149 DOI: 10.1016/j.stem.2012.12.013. ↩
- Tzifi F, Economopoulou C, Gourgiotis D, Ardavanis A, Papageorgiou S, and Scorilas A. The Role of BCL2 Family of Apoptosis Regulator Proteins in Acute and Chronic Leukemias. Adv Hematol. 2012;2012:524308. PMID: 21941553 DOI: 10.1155/2012/524308. ↩
- Chao DT and Korsmeyer SJ. BCL-2 family: regulators of cell death. Annu Rev Immunol. 1998;16:395-419. PMID: 9597135 DOI: 10.1146/annurev.immunol.16.1.395. ↩
- Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014;4:362-75. PMID: 24346116 DOI: 10.1158/2159-8290.CD-13-0609. ↩
- Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, Busman T, et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016;6:1106-1117. PMID: 27520294 DOI: 10.1158/2159-8290.CD-16-0313. ↩
- Chen F, Glytsou C, Zhou H, Narang S, Reyna DE, Lopez A et al. Targeting Mitochondrial Structure Sensitizes Acute Myeloid Leukemia to Venetoclax Treatment. Cancer Discov. 2019;9:890-909. PMID: 31048321 DOI: 10.1158/2159-8290.CD-19-0117. ↩
- Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell. 2015;163:1515-26. PMID: 26627737 DOI: 10.1016/j.cell.2015.11.015. ↩
- Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177-89. PMID: 16839885 DOI: 10.1016/j.cell.2006.06.025. ↩
- Burke PJ Mitochondria, Bioenergetics and Apoptosis in Cancer. Trends Cancer. 2017;3:857-870. PMID: 29198441 DOI: 10.1016/j.trecan.2017.10.006. ↩
- Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, Busman T, et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016;6:1106-1117. PMID: 27520294 DOI: 10.1158/2159-8290.CD-16-0313. ↩
- Wortel IMN, van der Meer LT, Kilberg MS, and van Leeuwen FN. Surviving Stress: Modulation of ATF4-Mediated Stress Responses in Normal and Malignant Cells. Trends Endocrinol Metab. 2017;28:794-806. PMID: 28797581 DOI: 10.1016/j.tem.2017.07.003. ↩
- Quiros PM, Prado MA, Zamboni N, D’Amico D, Williams RW, Finley D, et al. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J Cell Biol. 2017;216:2027-2045. PMID: 28566324 doi: 10.1083/jcb.201702058. ↩
- Tahir SK, Smith ML, Hessler P, Rapp LR, Idler KB, Park CH, et al. Potential mechanisms of resistance to venetoclax and strategies to circumvent it. BMC Cancer. 2017;17:399. PMID: 28578655 DOI: 10.1186/s12885-017-3383-5. ↩
- Bose P, Gandhi V, and Konopleva M. Pathways and mechanisms of venetoclax resistance. Leuk Lymphoma. 2017;58:1-17. PMID: 28140720 DOI: 10.1080/10428194.2017.1283032. ↩
- DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133:7-17. PMID: 30361262 DOI: 10.1182/blood-2018-08-868752. ↩