Lead-oriented synthesis: a new concept to aid drug-discovery process
Lead-oriented synthesis: a new concept to aid drug-discovery process
The discovery and development of new drugs is a long and expensive process, and despite of it, essential to face present and new diseases. For small molecules, which account for the majority of the marketed drugs, the discovery process generally involves finding a starting point termed hit or lead compound. These molecules have biological activity but need to be optimized to enhance their potency and selectivity (i.e. minimize the toxicity) and improve pharmacokinetic parameters making them suitable to go to the next stage, the pre-clinical tests.
Lead compounds are often found by screening hundreds of thousands of molecules from pharmaceuticals’ chemical libraries. In this rather random approach, scientist noticed that some molecules with a molecular weight (mw) of less than 500 Daltons (Da) and a LogP (a measure of lipophilicity) lower than 5 were more likely to end as marketed drugs 1 Together with constrains in the number of hydrogen bonding groups, they conform Lipinski’s rule of 5 which has historically been used to describe the small-molecule drug-like space. Further studies have refined the molecular properties that correlate with success in the drug-development process but molecular mass and specially lipophilicity, measured as cLogP (computed octanol-water partition coefficient) yet remain as the most important factors.
Lead optimization process almost inevitably produces heavier and more lipophilic molecules 2 as medicinal chemists add complexity and functionality to the molecule. Therefore, good chemical starting points that allow property inflation during optimization are essential, and as an analogy to the above mentioned drug-like space a lead- like space can also be defined (Figure 1). The concept of lead-oriented synthesis has recently been introduced 3 to emphasize the need of synthesizing small molecules which would lie in this space, this is, that would be good starting points for lead optimization.
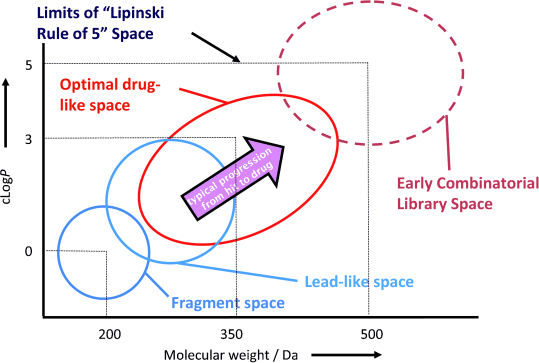
Indeed, this call is justified. Despite the vast amount of known compounds few meet the requirements to serve as the origin for drug development: more than 97% of commercially available molecules (approximately 4.9 million) are not lead-like. Moreover, modern synthetic chemistry fails too in this aspect, as most of the compounds reported in synthetic methodology papers do not comply with lead-likeness standards.
On top of that, the exploration of the chemical space has been uneven, biased by the fact that chemists are more likely to use a particular framework to make a compound if that framework has been used in the past. For example, in the case of heterocycles just 0,25% of the hetero frameworks are found in half of the compounds 4.
Obtaining molecules with desirable biological properties and suitable physicochemical parameters that ensure their efficacy inside the human body has always been the aim of drug-discovery programs. So far, the different approaches have been focused in obtaining drug-like molecules. This is the case of target oriented synthesis, which focuses on a molecule, designs the retrosynthetic analysis and then plans the synthesis. Its main drawback is that it just targets one compound, and having hundreds or even thousands of molecules to screen is preferred when new drugs are sought. On the other hand, combinatorial chemistry is ideal to produce a huge number of compounds, but often they have excessive molecular weight and/or lipophilicity. Diversity-oriented synthesis (fig 2) achieves structural diversity and complexity by the use of cascade reactions producing large number of molecular scaffolds using a small set of transformations. In the diversity-oriented synthesis structural diversity and complexity are sought, but the molecules obtained in this way are often large in size and lie in drug like space, so there is little room for optimization. These compounds might be useful as drugs themselves, but not as leads or hits. Fragment-based screening (fig 2) involves screening small molecules that are not intrinsically drug-like, but that might become subunits (fragments) of drug-like compounds.
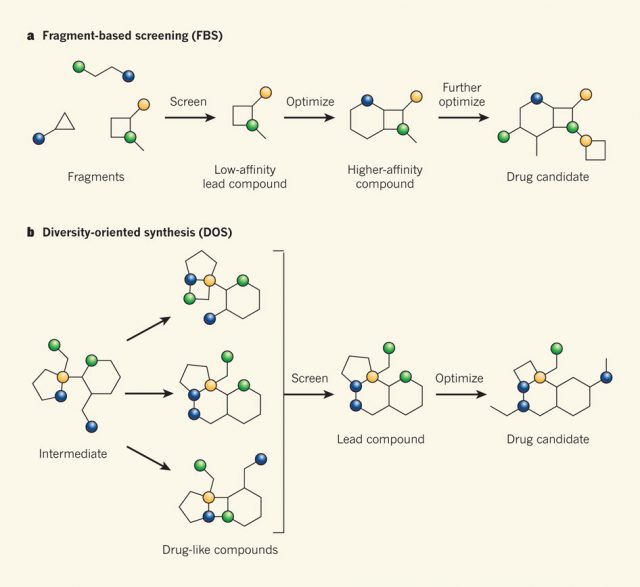
Through different approaches, the above mentioned methodologies can efficiently produce drug-like molecules, but they are limited to yield smaller molecules with the desired molecular properties (1
References
- C. A. Lipinski, F. Lombardo, B. W. Dominy, P.J. Feeney Adv. Drug Deliv. Rev. 1997,23, 3–25. DOI:10.1016/S0169-409X(00)00129-0 ↩
- M. Hann, G.M. Keserü Nat Rev Drug Discov2012, 11, 355-365. DOI:10.1038/nrd3701 ↩
- A. Nadin, C. Hattotuwagama, I. Churcher Angew. Chem. Int. Ed. 2012, 51, 1114 – 1122. DOI: 10.1002/anie.201105840 ↩
- A. H. Lipkus, Q. Yuan, K. A. Lucas, S. A. Funk, W. F. Bartelt, R. J. Schenck, A. J. Trippe J. Org. Chem., 2008, 73, 4443-4451. DOI: 10.1021/jo8001276 ↩
- Doveston R. & Adam Nelson (2013). Towards the realisation of lead-oriented synthesis, Drug Discovery Today, DOI: http://dx.doi.org/10.1016/j.drudis.2013.11.006 ↩
2 comments
this article is really new and very very important for me
[…] The discovery and development of new drugs is a long and expensive process, and despite of it, essential to face present and new diseases. For small molecules, which account for the majority of the marketed drugs, the discovery process […]