One kinase to rule diabetes? The role of JNK
One kinase to rule diabetes? The role of JNK

World Health Organization (WHO) established the recognition of obesity as a disease in 1948, but it was not until the last decade of the 20th century that people began to gain awareness of overweight-derived health problems. A decade ago and based on worldwide data indicating that 40 % of global adult population were overweight and 13 % were obese, WHO declared obesity a pandemic. Besides, obesity is an important risk factor of chronic diseases like type 2 diabetes and both conditions have shown a parallel increase worldwide. Due to this overwhelming increase, both obesity and type 2 diabetes represent significant challenges to social and healthcare systems across the globe.
Diabetes is a chronic disease that occurs as a result of unusually high glucose levels in the body system (hyperglycemia or what is commonly known as blood sugar). The main blood sugar regulator is the hormone insulin, which is produced by pancreatic cells when a raise in blood sugar is detected (going from de normoglycemia to hyperglycemia). Once it is secreted, insulin reaches body tissues (skeletal muscle, adipose tissue or liver) and tells them to capture glucose. Through this absorption, blood glucose levels go back to normal (normoglycemia).
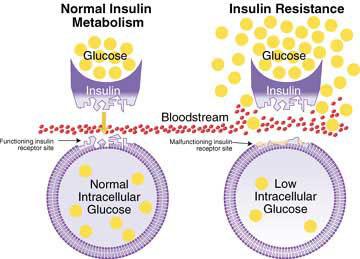
Particularly, in type 2 diabetes cells and tissues fail to respond normally to insulin, a condition known as insulin resistance. For instance, after every meal, blood glucose rises and pancreas secretes insulin, but as tissues ignore the hormone because they are insulin resistant, glucose is not absorbed and blood sugar levels remain high. In this context, pancreatic cells keep receiving signals to produce insulin because there is hyperglycemia. This leads to pancreatic failure, this is, the pancreas stops producing insulin and diabetes is eventually established. In this respect, type 2 diabetes tends to develop gradually, as the patient goes through a long insulin resistance phase before the pancreatic failure appears. Unlike type 1 diabetes (also known as insulin-dependent diabetes), it does not respond to insulin administration because cells show intrinsic resistance to this hormone. Hence, patients with type 2 diabetes are also treated with other drugs such as thiazolidinediones (TZDs), heterocyclic compounds that activate PPARγ nuclear receptor, which leads to alleviation of insulin resistance.
Dr. Caelles group studies the molecular mechanisms altered in type 2 diabetes and more specifically those involved in insulin resistance and triggered by JNK proteins.
JNK is a protein located within the cell and that has been proved to participate in many processes, principally in those leading to inflammatory response. In this sense, obesity–associated adipose tissue expansion is related to a low-intensity chronic inflammatory process, where JNK activity is atypically high. This increase of the JNK activity is one of the molecular causes of insulin resistance. Consequently, inhibition of JNK activity is associated to a better response to insulin and represents a potential therapeutic alternative for the treatment of type 2 diabetes. In this context, Dr. Caelles group proved 1 that TZDs prevent obesity-induced JNK activation through PPARγ and, consequently, they increase the response to insulin. From this study, it was concluded that TZD antidiabetic effect is based on this molecular mechanism.
Because TZDs receptor (PPAR) is mainly expressed in adipocytes, it was assumed that TZDs acted primarily on adipose tissue. Dr. Caelles then decided to study 2 whether TZDs could also act on insulin-secreting pancreatic β-cells, as TZDs receptor, although more discretely, is also expressed in those cells. In order to conduct this study, Dr. Caelles developed a transgenic mouse model that is also an example of how the activation of one sole protein in a specific cell type is able to modify homeostasis through the entire organism.
JNK needs to be activated in order to exert its role. Since the researchers wanted to focus in the role of JNK in insulin resistance in pancreatic β-cells, it had to be specifically activated in those cells. Hence, they chose a protein that activates JNK, known as MKK7, and more precisely they used a mutant version of the protein called MKK7D, whose presence in the cell keeps JNK activated. In order to conduct this study, MKK7D had to be expressed exclusively in pancreatic cells. For this purpose, a new line of transgenic mice was used. The mouse strain contained a transgene which expressed GFP (Green fluorescent protein) under the control of a constitutive promoter, so the mice would be fluorescent. This construct was injected in mice ovocytes and transgenic mice were obtained.
In order to have MKK7D expressed only in pancreatic β-cells, female transgenic mice were crossed with transgenic males containing another transgene coding for Cre-recombinase, a protein that targets Lox P signals (those flanking GFP gene). In these mice, Cre-recombinase gene expression is restricted to pancreatic β-cells, so GFP gene is eliminated, leading to the expression of MKK7D in this cell type. Finally, this enhances permanent and exclusive activation of JNK in pancreatic cells.
The study of these mice led to the conclusion that, even though JNK chronic activation does not affect either cell morphology or survival, nor insulin production, it does affect β-cells insulin secretion, which is triggered by hyperglycemia. Because of this, mice show glucose intolerance (this is, glycemia remains high for a longer period) and it is alleviated by TZDs. In addition to establishing the role of JNK in the regulation of insulin secretion in response to glucose, this work is a pioneer in proving the performance of this drug in pancreatic cells.
The transgenic mice generated in this work will be useful to continue with the study of molecular pathways in pancreatic cells and represent a valuable tool in diabetes research, a disease affecting 1 out to 11 people and responsible for the deaths of 1.5 a million people every year.
References
- Julieta Díaz-Delfín, Mònica Morales and Carme Caelles (2007) Hypoglycemic Action of Thiazolidinediones/Peroxisome Proliferator–Activated Receptor γ by Inhibition of the c-Jun NH2-Terminal Kinase Pathway Diabetes doi: 10.2337/db06-1293 ↩
- Jordi Lanuza-Masdeu, M. Isabel Arévalo, Cristina Vila, Albert Barberà, Ramon Gomis and Carme Caelles (2013) In Vivo JNK Activation in Pancreatic β-Cells Leads to Glucose Intolerance Caused by Insulin Resistance in Pancreas Diabetes doi: 10.2337/db12-1097 ↩
3 comments
[…] *Imagen de portada: Islote de Langerhans aislado de páncreas de rata. Los núcleos se muestran de color azul, mientras que las células beta están teñidas de color verde (insulina) y las células alfa de color rojo (Wikimedia Commons) *El […]
[…] Read the complete article… […]
[…] *Imagen de portada: Islote de Langerhans aislado de páncreas de rata. Los núcleos se muestran de color azul, mientras que las células beta están teñidas de color verde (insulina) y las células alfa de color rojo (Wikimedia Commons) *El […]