Avoiding geological timescales to access low energies in bulk glasses
Avoiding geological timescales to access low energies in bulk glasses

Common glass, used in windows or bottles, for example, is made by heating a mixture of calcium oxide (lime), sodium carbonate (soda), and silicon (IV) oxide (sand), resulting in a calcium silicate. This silicate is not a crystal but a solid in which atoms are positioned at random and have no long-range ordered pattern. These kind of solids, called “glasses”, characteristically have no definite melting point, softening over a range of temperatures instead. Thus, the so-called glass transition temperature is not a clear-cut temperature and the theory of the glass transition is much more complicated than the theory of normal phase transitions.
When dealing with polymers, we usually find a glass structure and behaviour; but if the polymeric units are big enough, their size fall in the nanometre range or, in other words, we have a glass that presents structure at the nano scale. This means that, even though it is still a glass, there is some kind of order at a considerable range. Actually, the alteration of glassy dynamics in nanostructured glasses has been one of the most intensely debated issues since the finding in the early 90s of depressed glass transition temperature in thin polymer films.
And it is so hotly debated, apart from its scientific interest, for its economic and technical importance. These nanostructured systems can be employed in a wide range of technologically relevant applications. For instance, submicron thick polymer films are used as templates in microelectronics, non-biofouling protective coatings, membranes in separation technologies, or active layers in photovoltaic cells. Polymer nanocomposites are extensively employed in tire technologies and in those applications where mechanical reinforcement, enhancement of barrier properties, flame resistance, electro-optical, and bactericidal properties, are required. Polymer nanospheres have received increasing interest in the last years in applications such as vehicles in drug delivery, components in fluorescent imaging, performance reinforcing additives, and components in photonic structures, to name just a few.
The point is that understanding vitrification in glass-forming systems still remains an unsolved important problem in condensed matter physics. This is the case because the glassy state is a metastable state, with the transition temperature (Tg) becoming lower as the rate of cooling decreases.
The conventional wisdom assumes that vitrification takes place when the timescale related to the molecular mobility, defined by the time to get to the equilibrium or relaxation time, τ, is of the order of the inverse of the cooling rate, βc. Below Tg, the nonequilibrium glass spontaneously evolves toward equilibrium, a phenomenon known as structural recovery or physical aging. Within the conventional wisdom on the glass transition, such recovery is exclusively related to τ. Importantly, from a conceptual point of view, vitrification and molecular mobility are different aspects of glassy dynamics. The former entails information on how equilibrium is lost when we cool a melt in equilibrium or recovered in the glassy state.
The thermodynamic state attained after vitrification or after aging in some set of conditions is commonly quantified using the concept of the fictive temperature, Tf , defined as the temperature at which a glass in a certain thermodynamic state would be at equilibrium. Probing the molecular mobility requires the application of small perturbations – in particular, smaller than the amplitude of spontaneous fluctuations – conducted at invariant Tf.
This is correct if we are considering bulk experiments. But experiments and simulations on glasses geometrically confined at the nanoscale have revealed a decoupling between glass transition and physical aging on the one hand, and molecular mobility on the other. In particular, for polymeric glasses exhibiting a large amount of free interfacial area, negative Tg deviations can be observed, even in the presence of essentially bulk-like molecular mobility.
Another aspect of glassy dynamics challenging the conventional wisdom regards the existence of different equilibration mechanisms in the glassy state. In contrast to the standard behaviour based on a single decay toward equilibrium for glasses aged close to Tg after relatively small temperature jumps, recent experiments in a wide variety of glasses showed that two or more mechanisms of equilibrium recovery can be identified, provided that these experiments are performed substantially below Tg and over timescales often exceeding several weeks. The presence of different mechanisms of equilibration is directly correlated to intermediate metastable states. From the viewpoint of the timescales involved in glass equilibration, the slowest mechanism of equilibration exhibits the temperature dependence generally associated with the glass transition, while the fast mechanisms generally display a mild temperature dependence.
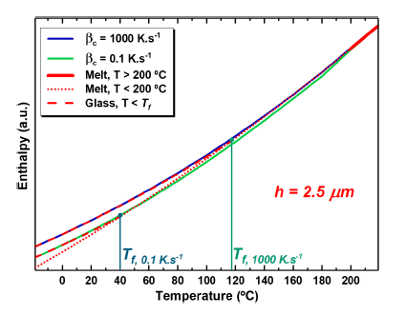
with a characteristic length scale of 2.5 μm on heating at
1000 Ks−1 after cooling at 1000 Ks−1 (blue line) and 0.1 Ks−1
(green line). The melt enthalpy (red dotted line) and the glass enthalpy (red dashed lines) are also shown.
In this context, Xavier Monnier (DIPC) and Daniele Cangialosi (CFM & DIPC) have used 1 fast scanning calorimetry to investigate the effect of size on the glass transition and molecular mobility of poly(4-tert-butylstyrene) (PtBS), a polymer that exhibits large confinement effects in terms of Tg depression.
The samples had a typical size in the micrometer scale, well beyond the length scale at which confinement effects on the glass transition are generally found. The researchers characterised the glass transition is characterized by assessing the limiting Tf, that is, the temperature the system would be at equilibrium after cooling at a given rate.
The results showed tremendous Tf negative deviations, as large as 80 K for the smallest sample, from bulk behaviour. These deviations were found to be largely associated with a fast mechanism of equilibration. In contrast, molecular mobility exhibited bulk-like behaviour irrespective of the sample size.
Such huge deviations indicate that accessing thermodynamic states with low Tf is strongly facilitated by the presence of a large amount of free interface. The main consequence is that we have now a formidable means to attack the challenges that the study of glass transition present, avoiding the geological timescales required to access low energies in bulk glasses.
Author: César Tomé López is a science writer and the editor of Mapping Ignorance.
References
- Xavier Monnier and Daniele Cangialosi (2018) Thermodynamic Ultrastability of a Polymer Glass Confined at the Micrometer Length Scale Physical Review Letters doi: 10.1103/PhysRevLett.121.137801 ↩