Tuning the excited-state Hückel‐Baird hybrid aromaticity
Tuning the excited-state Hückel‐Baird hybrid aromaticity
Molecules when excited from their ground state (S0) to their lowest electronically excited states often change their electronic structure considerably, which impacts on a range of important molecular properties. For example, the reactivity of a molecule in its excited state often differs markedly from that in its S0state. Also, the charge distribution within a molecule is normally altered upon excitation, and consequently, its interaction with the surrounding medium.
The electronic structure changes upon excitation are influenced by a number of factors; those that are intrinsic to the molecule and those that are extrinsic. The bonding and antibonding features of the orbitals populated in the excited state is one of the most obvious intrinsic factors, whereas the polarity of the surrounding solvent or medium (for example, the active site within a protein) constitutes a typical example of possible extrinsic factors. An important intrinsic factor is the ability of a molecule to switch its electronic structure from a pro-aromatic structure to an aromatic one upon excitation.
Aromaticity is a property of cyclic (ring-shaped), planar (flat) structures with pi bonds in resonance (those containing delocalized electrons) that gives increased stability compared to other geometric or connective arrangements with the same set of atoms. Aromatic rings are very stable and do not break apart easily.
An aromatic ring contains a set of covalently bound atoms with specific characteristics:
- A delocalized conjugated π system, most commonly an arrangement of alternating single and double bonds
- Coplanar structure, with all the contributing atoms in the same plane
- Contributing atoms arranged in one or more rings
- A number of π delocalized electrons that is even, but not a multiple of 4. That is, 4n+2 π-electrons, where n = 0, 1, 2, 3, and so on. This is known as Hückel’s rule.
According to Hückel’s rule, if a molecule has 4n+2 π-electrons, it is aromatic, but if it has 4n π-electrons and has the first three characteristics listed above, the molecule is said to be antiaromatic. The aromatic character of a molecule in the ground state can be verified experimentally using nuclear magnetic resonance, as this technique would detect a ring current in aromatic compounds.
This is the standard definition of aromaticity. But it is not that simple. The lowest triplet state of an annulene is, according to another rule, Baird’s rule, aromatic when it has 4n π-electrons and antiaromatic when the π-electron count is 4n + 2, where n is any positive integer. This trend is opposite to that predicted by Hückel’s rule for the ground state, which is usually the lowest singlet state. Through various theoretical investigations, this rule has also been found to extend to the lowest lying singlet excited state (S1) of small annulenes.
Baird’s rule has thus become known as the photochemical analogue of Hückel’s rule. The Baird-aromaticity concept be developed into a tool that is useful to the design of molecules with targeted optoelectronic properties for potential use in, for instance, organic electronics. But a correct interpretation of its experimental signature is crucial for that development.
Thus, research on excited state Baird-aromaticity has intensified in the last decade. Comprehensive and critical analyses combined with solid computational assessments has been used for the proper interpretation of experimental data as there is presently no spectroscopic technique that can be used as the sole method to evaluate excited state aromaticity in a similar manner as NMR spectroscopy can be used to assess aromaticity in the S0state.
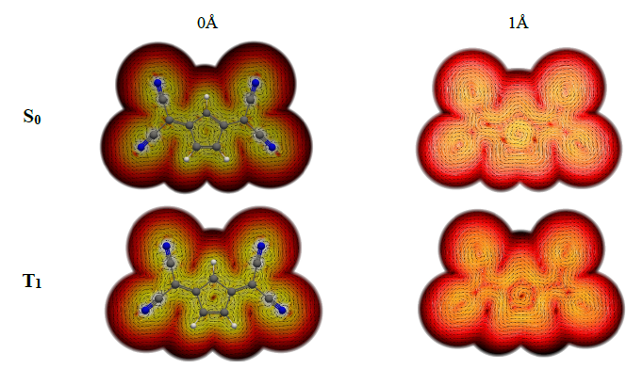
Pro-aromatic molecules are molecules that in a higher-energy state are significantly influenced by resonance structures in which conjugated rings take on Hückel-aromatic character. It has been argued in recent studies that there are also pro-aromatic molecules that adopt central units with 4np-electron Baird-aromatic character in their lowest triplet diradical states (T1), although detailed analysis suggests that these compounds are better labelled as T1Hückel-Baird hybrid molecules, where Hückel-aromaticity is the dominant form.
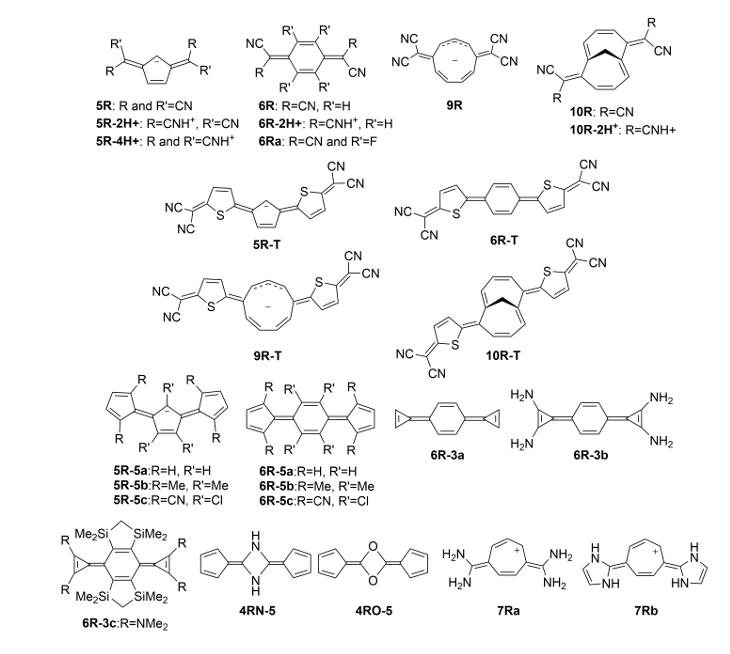
Now, a team of researchers has considered 1 a series of symmetrically substituted conjugated rings with potential Baird aromaticity in the lowest-lying excited triplet and singlet states (Figure 2). Its results allow to establish general guidelines for the rational design of molecules with excited state Hückel/Baird aromaticity in pro-aromatic quinoidal compounds.
The authors conclude that the characterization of Baird aromaticity needs to be carried out with care and that the labelling of a conjugated ring as Baird aromatic requires a detailed analysis of the excited state electronic structure. They suggest a safe procedure in which several parameters and computational tools are combined, a newly introduced energy-based analysis of the Hückel and Baird configurations among them.
The new analysis indicates that the symmetric substitution of conjugated rings favours the presence of low-energy excited states with small changes in the number of p-electrons in the central conjugated ring.
The researchers found two main strategies to promote a high Baird aromatic character of the central ring, by using either anionic and small conjugated rings with electron donating groups as substituents and small exocyclic groups with electron withdrawing substituents, or electron deficient conjugated rings with exocyclic electron-donor substitution.
Author: César Tomé López is a science writer and the editor of Mapping Ignorance
Disclaimer: Parts of this article may be copied verbatim or almost verbatim from the referenced research papers.
References
- Sílvia Escayola, Claire Tonnelé, Eduard Matito, Albert Poater, Henrik Ottosson, Miquel Solà, David Casanova (2021) Guidelines for Tuning the Excited State Hückel‐Baird Hybrid Aromatic Character of Pro‐Aromatic Quinoidal Compounds Angewandte Chemie doi: 10.1002/anie.202100261 ↩