Donostia Natural Orbital Functional (DoNOF), an open-source program for quantum chemistry
Donostia Natural Orbital Functional (DoNOF), an open-source program for quantum chemistry
Today, computational chemistry helps the experimental chemist understand experimental data, explore reaction mechanisms or predict completely new molecules. There is no doubt that understanding at the molecular level will ultimately lead to an ab initio process design. Thus, the solution of the quantum mechanical many-electron problem is one of the central problems of physics and far from trivial. The best solutions to this problem are offered by methods based on approximate wave functions, however, such techniques demand significant computational resources as soon as the systems grow.
The solution of the quantum mechanical many-electron problem is one of the central problems of physics and far from trivial. Probably the most popular method at present is density functional theory (DFT). DFT is a theoretical treatment of molecules in which the electron density is considered rather than the wave functions of individual electrons. In other words, a way of describing many-electron, in general, many-fermion, systems in which the energy is a functional of the density of electrons (fermions).
DFT is based on the Hohenberg-Kohn theorem which asserts that the electronic charge density completely determines a many-electron system and that, in particular, the total energy is a functional of the charge density. In other words, it considers a electron density in the molecule so that the system behaves exactly as if the electrons were spread out into a continuously distributed charge, instead of the wave functions of individual atoms.
Nowadays, we have numerous empirical and non-empirical parametrized functionals, but this large-scale parametrization has led to a very specific applicability in systems and phenomena. For each new challenge, density functionals have to be calibrated against the standard wave function methods. The latest proposed strategies represent steps forward in terms of accuracy, but they also imply higher computational costs.
What is needed is a one-particle formalism more accurate than approximate density functionals, but less computationally demanding than wave function methods. The one-particle reduced-density matrix of the ensemble of pure-spin states provides the proper description of spin multiplets. The answer may be to develop a functional based on this reduced matrix.
In 2017, Mario Piris, an Ikerbasque research professor working at DIPC and UPV/EHU, proposed a global method to calculate electron correlations from a different perspective, based on so-called natural orbitals. Natural orbitals are defined as eigenfunctions of the spinless reduced electron density matrix, a matrix that appears when you treat the system and its electron wave functions in linear-algebraic terms, in what is known as Hartree-Fock aproximation. The natural orbital functional theory (NOFT) has emerged as an alternative formalism to both density functional and wave function methods.
In NOFT, the electronic structure is described in terms of the natural orbitals and their occupation numbers. The approximate natural orbital functionals have proven to be more accurate than those of the density for systems with a significant multiconfigurational character, on one side, and scale better with the number of basis functions than correlated wave function methods, on the other side.
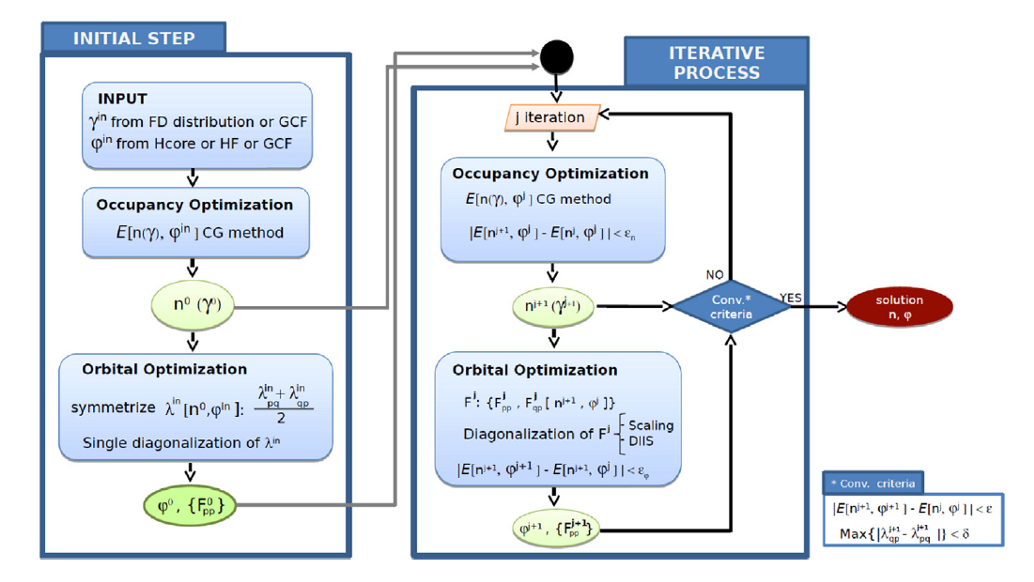
Now, Piris and Ion Mitxelena (DIPC) present DoNOF (Donostia Natural Orbital Functional) 1, their open source implementation based on diagonalizations that allows to obtain the resulting orbitals automatically orthogonal. The DoNOF computer program is designed to solve the energy minimization problem of a natural orbital functional that describes the ground-state of an N-electron system at absolute zero temperature.
The capabilities of DoNOF were put to the test with the water molecule. This included analysis of molecular orbitals, ionization potential, electric moments, optimized geometry, harmonic vibrational frequencies, binding energy, and potential energy surface of water symmetric dissociation. DoNOF showed that it can calculate the energy and a variety of properties of any isolated molecule in its ground state, regardless of the total spin value of the system.
Interestingly, no prior knowledge of the system is required, so the method can be employed as a black box. In addition, the electron-pair-based natural orbital functional is parameter free, so the performance is not subject to specific systems or parametrization data.
DoNOF is written in Fortran and published under a GPLv3 license. DoNOF is available from the following links:
Author: César Tomé López is a science writer and the editor of Mapping Ignorance.
Disclaimer: Parts of this article might have been copied verbatim or almost verbatim from the referenced research paper.
References
- Mario Piris & Ion Mitxelena (2020) DoNOF: An open-source implementation of natural-orbital-functional-based methods for quantum chemistry Computer Physics Communications doi: https://doi.org/10.1016/j.cpc.2020.107651 ↩