Williams syndrome and autism in the same child
Williams syndrome and autism in the same child
Williams syndrome (WS) was first described in 1961 by New Zealand cardiologist John Williams. It is mostly caused by the microdeletion 7q11.23, a loss of a fragment of genetic material on chromosome 7. It is usually a “de novo” mutation, that is, it is not inherited from the parents, but rather arises in that child spontaneously.
Among the genes lost in this region is ELN, which encodes elastin, a protein that, as its name indicates, is involved in the levels of elasticity of different organs. We need elasticity in different organs, such as the arteries that dilate with the heartbeat and return elastically to their initial size. The reduced production of this protein causes problems in the arteries and in the heart, such as supravalvular aortic stenosis, which is common in patients with Williams syndrome and which explains why the syndrome was first identified by a cardiologist.
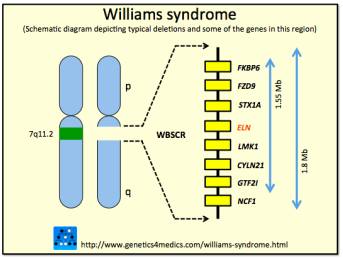
This chromosomal alteration has systemic effects, leads to a global delay in development, and causes various symptoms and signs. A much-commented characteristic is that these children have a special face: a flattened nasal bridge, a shorter nose, a larger frontal region, a thinner chin, thicker lips and a larger mouth. Colloquially, these children are said to have the face of a leprechaun, which has been called the elf face. In terms of neurocognitive and neuropsychomotor development, children with WS usually show a lower than average IQ, ranging from 60-70, which constitutes a mild to moderate intellectual disability. They also tend to present hypotonia in the extremities and the consequent general delay in motor development. Finally, significant difficulties have also been found in these children in relation to visual and spatial cognition. However, many studies have shown that the processing of facial gestures and language are preserved in this population, although this finding is a controversial subject in the literature.
An almost universal fact among WS patients is their great interest in music, despite not being able to tolerate high-intensity noise. Another fundamental characteristic of children with WS is their hypersociability. They cannot stop hugging people, are not afraid of strangers, and love and trust everyone equally. They treat someone they just met the same way as a close person they have been living with for years. It is not easy, neither for them nor for their parents, nor for anyone else. That surrender, that trust can be dangerous, it makes them vulnerable, it makes them aliens to other children.
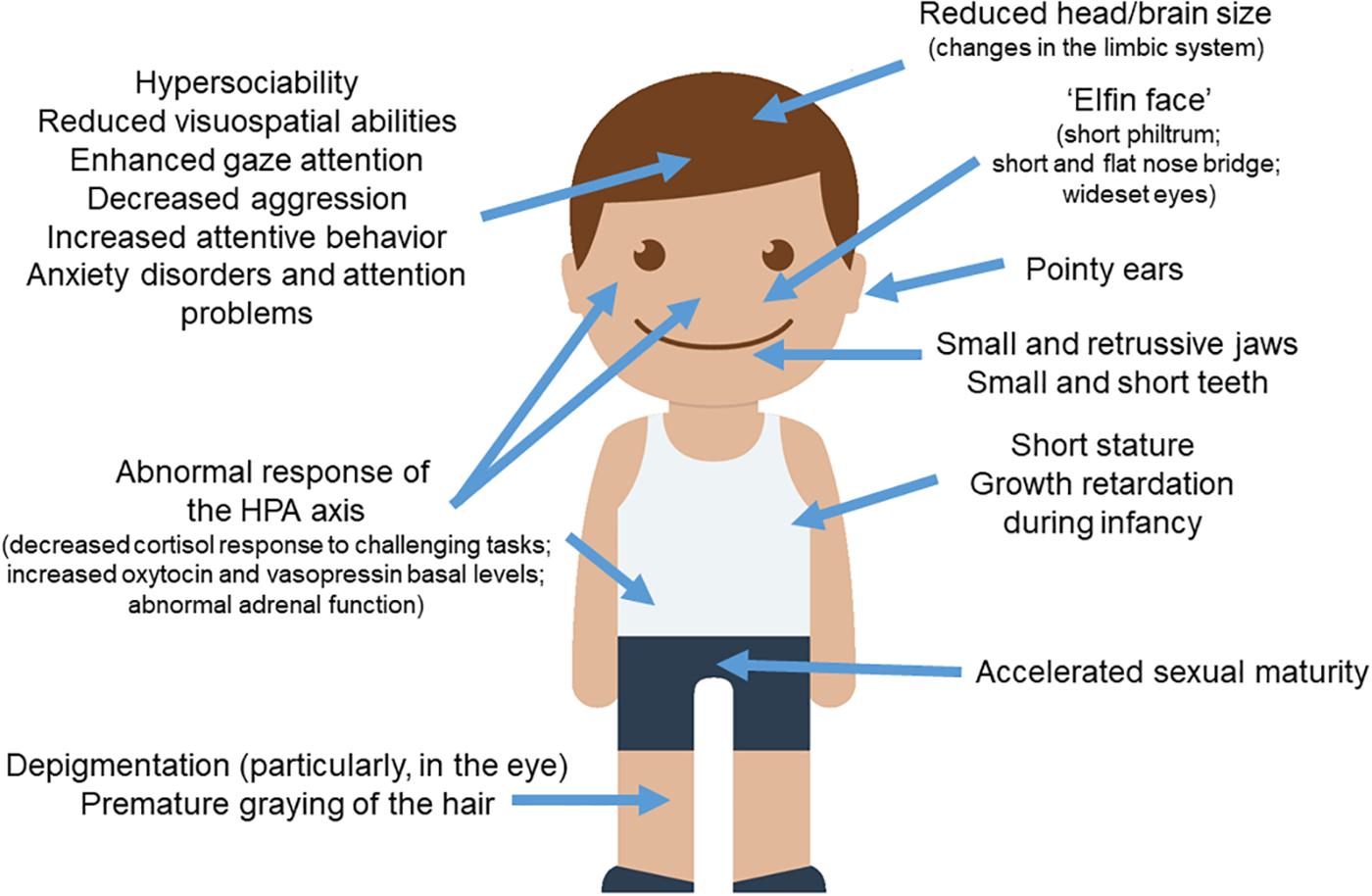
In a way, Williams syndrome is considered the opposite of autism. If children with WS are hypersociable, children with autism spectrum disorder (ASD) often have clear difficulties in socializing. ASD is a neurodevelopmental disorder characterized by two main signs: deficits in social interactions and communication, and repetitive or stereotyped behaviors. It is now accepted that a combination of genetic and environmental factors is what leads to the development of ASD in an individual. As in the case of WS there are many of these mutations that appear de novo, which are not inherited from either the father or the mother.
Carvalho and Mazza 1 have presented the case of a child, H., who presents both autism and WS. H. is African-American and is four years old. He was referred to the neuropediatric department of a university hospital by the genetics department after microarray testing gave a positive diagnosis of WS. His mother reported developmental delays in her child, along with walking and language difficulties. H. had been born by caesarean section due to ultrasound abnormalities detected during pregnancy that showed poorer than normal growth. H. was born when her mother was 34 years old, and he has an older brother. H.’s mother and brother are healthy, but his father has bipolar disorder. The mother also stated that H.’s father’s family has many members with psychiatric and behavioral disorders.
At the pediatric clinic, the following details were noted: the child was beginning to learn to speak, but showed an obvious delay in speech development; he had no sphincter control and was very irritable. As far as social interactions are concerned, he was able to engage in conversations with other people, but he was not fluent. He also often hit his head on the floor and slapped himself when he was upset. H. also could not eat alone, as he had difficulty chewing.
During the consultations, the child showed mood swings. At first, he was euphoric and seemed interested in new people and objects, with more attention to the latter. At other times, he was very irritable and not very communicative, exhibiting the self-injurious behaviors already described, and did not allow the physical examination. He was calmed when certain songs were played in the room.
In a follow-up visit, the child had recently abandoned early stimulation and had acquired a great and notable development in language, with a notable expansion of his vocabulary; his language had become more articulate and he was beginning to form sentences with more than three words. However, this language was limited to being instrumental; that is, he preferred to speak for more pragmatic and immediate purposes, like asking for a glass of water. Some of his expressions remained incomprehensible and had some vocal stereotypes. He had started going to school and daycare and had very little intimacy with his classmates or teachers, although he showed no conflict while with them. As far as daily life activities were concerned, he had significant deficits. His most frequent stereotype was that of spinning on his own axis. Initially he also had the habit of tiptoeing, but he had abandoned it. In general, he showed an irregular social smile and preferred to spend his time with inanimate objects for the most part, in most cases not showing interest in strangers in the clinic, with some exceptions, as described above. At the time of the physical examination, he showed extreme resistance to being touched and initiated self-harming behavior.
In terms of neuropsychomotor development, he had begun to sit without support at eight months and had begun to pick up objects at the age of one year; he had begun to walk only at one year and seven months. When he reached his current age of four, he began to point out things he wanted or was interested in; but he still did not make any gestures and depended on the instrumental use of adults when he wished something. His gait was rather ataxic, with static and dynamic imbalance, as well as the presence of hypotonia in his upper and lower limbs.
As for complementary tests, the electroencephalogram was normal for his age. The audiometry, on the other hand, was affected by the patient’s refusal to put on the headphones; therefore, the test was performed in a free field, but no significant hearing loss was initially detected.
The joint appearance – and diagnosis – of WS and ASD is still a very controversial issue. Quite a few authors commonly describe the two disorders as being in opposition to each other, mainly because the behavioral and social profile of children with ASD is that of very outgoing, friendly, and affable individuals who have no difficulty initiating interactions with others. However, there is evidence that WS patients who are fluent speakers have some symptoms associated with ASD, especially related to verbal, communication, and language development. Despite this, children who are learning to speak seem to have greater difficulty, indicating that a diagnosis of ASD may require some patience.
References
- Carvalho MM, Mazza J (2020) Williams Syndrome Associated With Autism Spectrum Disorder: A Case Report and Review of the Literature Cureus 12(8): e9641. doi: 10.7759/cureus.9641 ↩
2 comments
[…] Neurologiaren ikuspuntutik autismoaren kontrakoa da Williams sindromea. Biak dituen ume baten kasua topatu da, hala ere. J.R. Alonsoren Williams syndrome and autism in the same child […]
can symptoms that occur spontaneously occur at any age of the child?