Mimicking nature to face the green environmental transition
Mimicking nature to face the green environmental transition
“Any being, if it varies however slightly in any manner profitable to itself… will have a better chance of surviving, and thus be naturally selected. From the strong principle of inheritance, any selected variety will tend to propagate its new and modified form.”
― Charles Robert Darwin (1809-1882), The origin of species, 1860
Since the outset of industrial era, anthropogenic activities have stressed continuously the ecosystems. The hectic pace of technological advances during the XX and XXI centuries and the increasingly environmental pollution are flip sides of the same coin. Although nature has always found smart and sophisticated ways to palliate environmental imbalances, the rate of production and release of harmful substances by humankind is out of step when compared to the bio-machinery self-adaptation.1 As a consequence, the accumulation of greenhouse gases and persistent pollutants have disturbed the equilibrium of atmospheric, soil and water cycles. Therefore, the shift to a zero-toxic environment needs to be accompanied not only by the removal of these harmful substances from the environment, but by the green production of biofuels in order to reduce the carbon fingerprint of the human activities.2
Nature faces environmental pollution by trapping and transforming harmful chemicals and gases into bio-useful substances. Within the complex bio-machinery able to deploy this function, enzymes, and specifically, metalloenzymes, have predominant roles.3,4 Though bioengineering direct evolution has rapidly adapted enzymes and metalloenzymes to industrial applications, these biosystems collide with an insurmountable barrier: the lack of chemical and thermal stability to function out of the usual window where life is possible. This is mainly because the beauty and complexity of the three-dimensional structures of enzymes and proteins resembles on weak interactions that break down when exposed to chemically or thermally stringent conditions.5
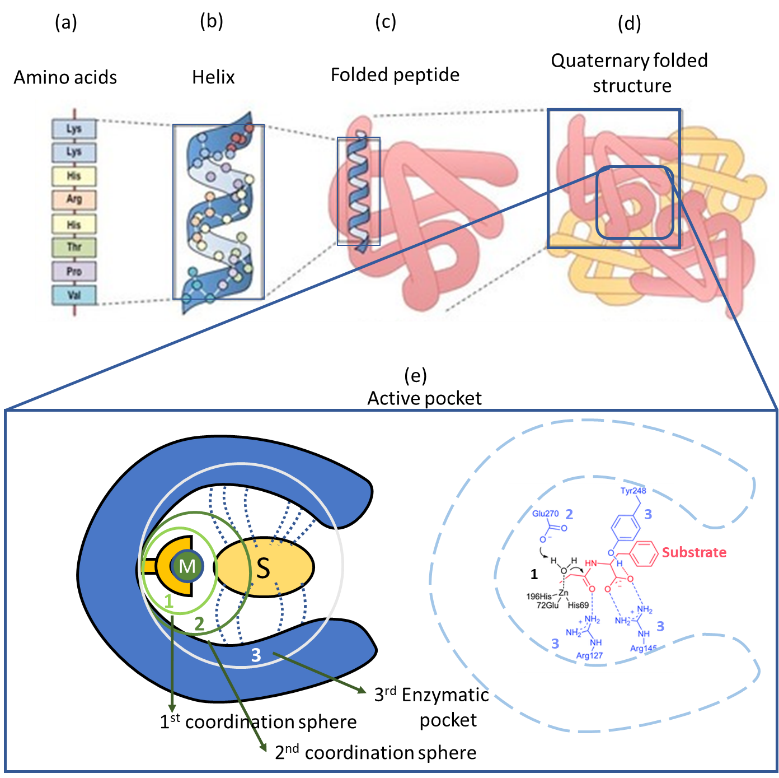
Enzymes are complex three-dimensional structures build up from the folding of long amino acid chains, also called peptides (Figure 1a-c). Therefore, the flexible and dynamic scaffolds of enzymes are mainly shaped by the weak interactions established by amino acids’ residues within the peptide sequences (Figure 1d). The structure is further stabilized and decorated with water molecules, prosthetic groups and cofactors hosted within the enzyme; which in addition play critical structural and functional roles (Figure 1e).
The versatility of enzymes has evolved to shape outstandingly sophisticated active-pockets able to recognize and transform single molecules by means of key-lock mechanisms. In the specific case of metalloproteins, the active pocket allocates single or clustered metal ions that are coordinated to the amino acid residues coming from the enzyme’s scaffold. Generally speaking, the bio-metals hosted within these complex structures possess strained coordination spheres that are usually completed with labile species (i.e. water or amino acid binding groups). Upon removal of the labile coordinating groups, metalloenzyme core is activated giving rise to highly reactive open catalytic sites (Figure 1e).6
But not only the first metal coordination shell matters. The specific location and disposition of amino acid residual groups, disposed at the second pseudo-coordination distances, fine-tunes the bio-metals catalytic activity by altering their electronic and redox properties. In parallel, this second shell also modulates the electric field and hydrophobic/hydrophilic nature of the enzymatic pocket that surrounds the metal core. The combination of the surface decoration of the pocket and the plasticity of the overall structure help to specifically adapt its shape and function to recognize, activate and transform the hosting substrates (Figure 1e).7
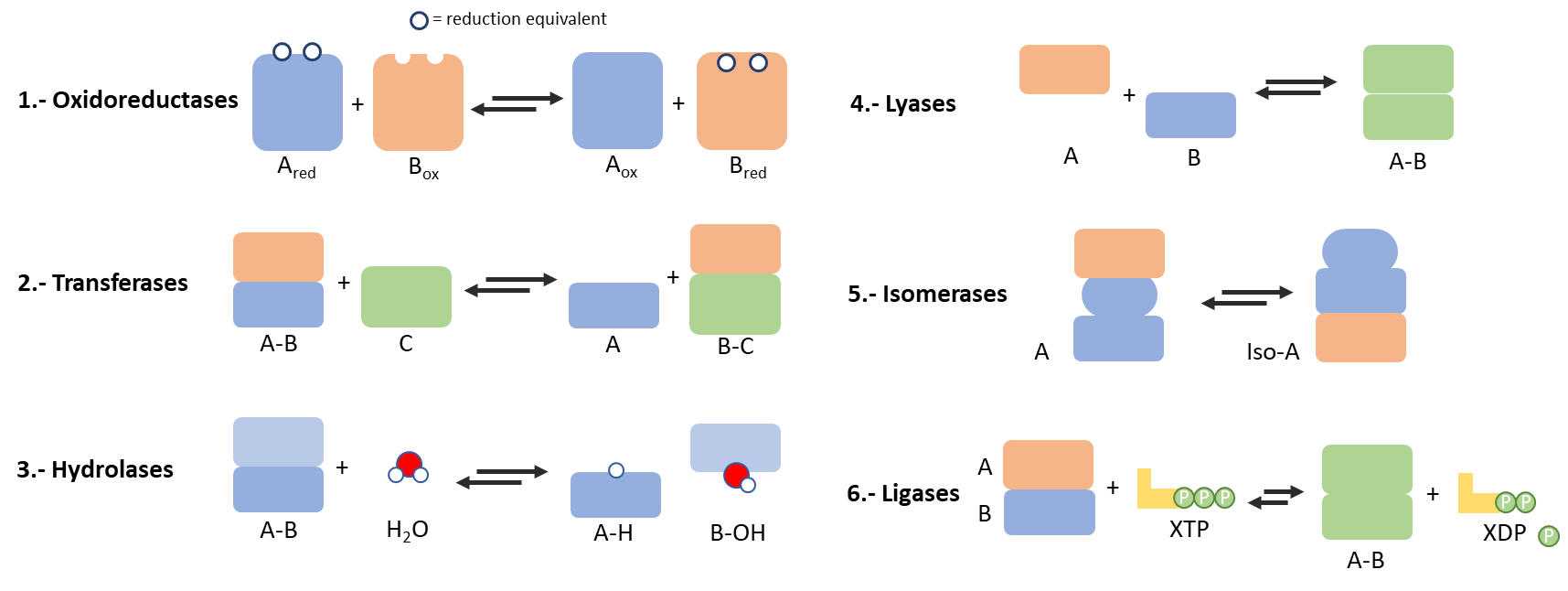
Enzymes and metalloenzymes are classified in function of their individual functionalities in six main groups: oxidoreductases (Fe, Cu and Mo ions), transferases (Mg, Mn and Co metals), hydrolases (Ca, Zn, Co, Mn and Mg ions), lyases (mainly based on Ca, Mg, Mn, Co and Zn), isomerases (Mn, Co and Fe biometals) and ligases (Mg and Mn ions) (Figure 2). By encompassing their individual functions, enzymes participate in such beautiful and sophisticated processes as photosynthesis or ion/electron tunneling transportation and regulation. These are just a couple of the multiple examples that require an enzymatic activity coupled in a self-regulated and reversible cascade mode, where the output of one enzymatic activity (i.e. light, electron, protons of organic molecules) feeds the input of the following one.8
Intense research efforts have been deployed to expand the metalloprotein biochemistry in order to obtain robust systems able to work out of the narrow window where the unfolding of the enzymes occurs. On one hand, the biochemists have been able to expand the application window of enzymes for industrial and environmental purposes, but their stability is still limited to soft temperatures, acidity and solvent conditions.9 In addition, the immobilization and/or encapsulation of enzymes and metalloenzymes in robust scaffolds has opened the perspective to further improve their long-term stability.10 On the other hand, many researchers have explored the assembly of homogeneous catalysts based on coordination compounds, which resemble oversimplified metal-core structure of the enzymes. But still, as homogeneous catalysts, these coordination compounds show similar limitations to those of the enzymes themselves. In a third approach, the research community has tried to replicate the metalloenzymes´ active cores in many polymeric, biopolymeric and inorganic supports, although the control over the second coordination shell of the bio-metals, as well as over the shape and chemistry of the catalytic pockets still remains a challenge.11
A question arises at this point, can the research community mimic metalloenzyme functions while improving their stability when their specificity and effectiveness rely on flexible three-dimensional structures stabilized by weak interactions?
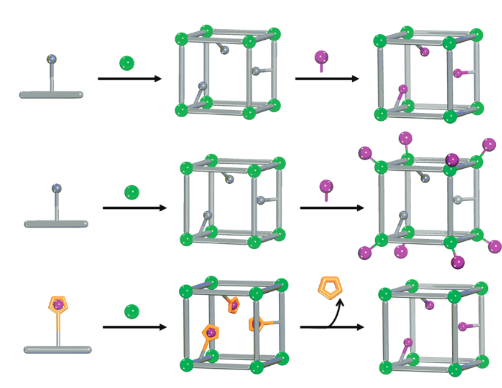
Here is where Metal-Organic Frameworks (MOFs) come into play. MOFs are highly porous and ordered materials built up from metal oxo-clusters and organic linkers connected through strong covalent bridges.12 Depending on the connectivity, geometry and length of the clusters and linkers´ building blocks, the final topology, porosity and pore chemistry of MOF materials can be fine-tuned to approach the metalloenzyme biochemistry. For instance, to date reticular chemistry has enabled 1) building up MOF materials from bio-metal and/or bio-organic constructs (i.e. peptides, aminoacids, porphyrins…) resembling the pockets and/or prosthetic groups of certain metalloenzymes,13 2) exchanging or attaching metal ions within the framework to approach the metal-ligand binding modes of metalloenzymes pockets, and 3) incorporating amino-acid residues as functional struts to further approach and tune the artificial metalloenzyme functions (Figure 3).14 In addition, in spite of their highly robust frameworks, some MOFs exhibit astoundingly flexible structures able to re-adapt their overall porosity and pore aperture depending on the host allocated within the pores. Similar to 3D flexible scaffold of enzymes, some MOFs can approach host molecules recognition mechanisms by distorting and readapting their frameworks.15
At this stage, most of the research carried out in MOF based metalloenzymes have been focused on the construction or installation of metal cores similar to the ones found in biocatalyst. Some efforts have been also carried out to tune the coordination modes of the metal ions installed within the MOFs. Even though we are in a nascent stage of this research approach, the initial results are really promising, demonstrating the potential of these systems for green fuels production and environmental remediation purposes. Three are the main applications areas where MOF-metalloenzyme systems have been assessed up to date:
-
Global decarbonization through bienzymatic anhydrase-like activity: carbon dioxide capture and transformation to carbonate (Type III hydrolase activity).
-
Green fuel production mimicking monooxygenase and hydrogenase type enzymes: proton reduction to produce hydrogen / catalytic oxidation of alkanes (Type I oxidoreductase enzymatic activity).
-
Oxidation and inactivation of highly hazardous chemicals as phenols, pesticides and warfare agents (Type I oxidoreductase and Type III hydrolase activities).
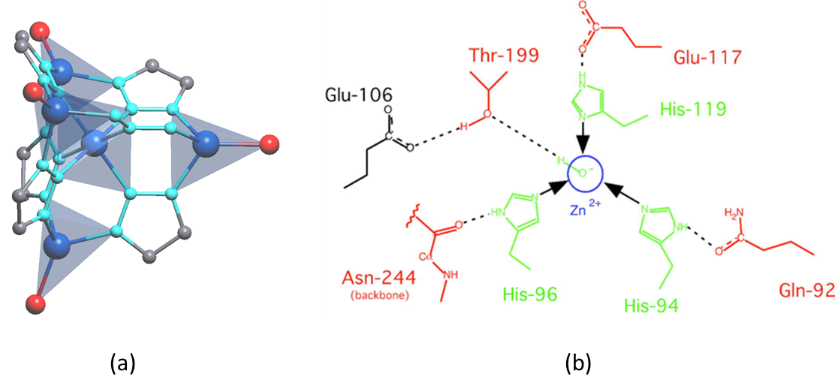
A remarkable example of a MOF with enzymatic activity over the capture and transformation of carbon dioxide was reported by M. Dincă and co-workers.16 In order to design a MOF with carbonic anhydrase (CA)-like activity they rationally modified the metal-nodes of the well-known MFU-4l MOF (terminal N3Zn‒Cl units converted into N3Zn‒OH units) (Figure 4). By following this approach, they increase its resemblance to carbonic anhydrases-like enzymes, which catalyse the hydration and dehydration of carbon dioxide (H2O + CO2 ↔ HCO3‒ + H+). The resulting material adsorbs high quantity of CO2 in a reversible manner (N3Zn‒OH ↔ N3Zn‒OCOOH), but in parallel, it is also active for the hydrolysis of 4-nitrophenyl acetate, mimicking the activity of CA. Given the significant uptake capacity (3.41 mmol g-1 at 298 K) and limited energy cost required for regeneration, this material gives great promise for CO2 capture and sequestration.
Direct conversion of alkanes to easily condensable oxygenated energy carriers (methanol, ethanol…) is conceptually a key reaction in the decentralized conversion of shale gas. While methane can be converted to methanol at very high temperatures, direct and highly selective methane oxidation pathway to methanol at ambient temperatures is a matter of metalloenzymes.
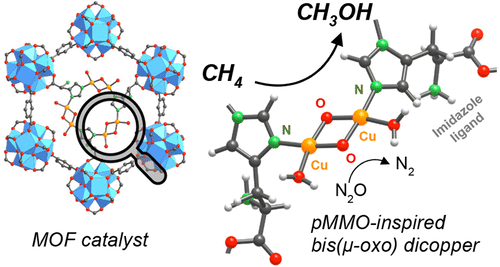
Alkanes oxidation at low temperatures path has also been proven to be feasible in metalloenzyme type MOFs. Starting from MOFs with metal-clusters resembling the metal-oxo moieties found in iron-oxidases, D. J. Xiao and co-workers proved that high spin iron ions of Fe2(dobdc) MOF are able to transform ethane to ethanol at 75% with a 25:1 selectivity over acetaldehyde. Iron open metal sites in Fe2(dobdc) work as reactive centres to activate N2O and drive the posterior oxidation of ethane to ethanol.17 Similarly, several micro and mesoporous zirconium-based MOFs have served as hosting matrixes for the installation of monomeric, dimeric and trimeric copper and iron species able to deploy methane monooxygenase type activity at 85 ºC.18,19 Going a step further, J. Baek and coworkers were able to install histidine residues coordinated to dimeric copper units within the zirconium trimesate MOF-808 (Figure 5). In an arrangement close to that observed in copper methane monooxygenase, the material showed the highest reported methanol productivity and selectivity (71.8 µmol/g) at isothermal treatments at 150°C.20
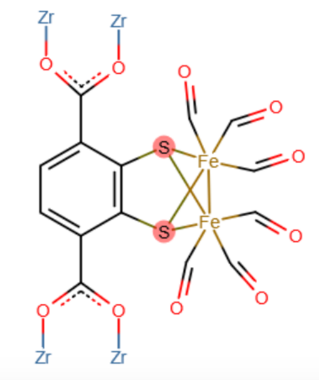
In the context of light-to-fuel conversion schemes, FeFe binuclear complexes have also been immobilized in zirconium MOF matrixes for the green production of hydrogen. The FeFe hydrogenase-like site has been stabilized within the porous framework of the UiO-66 zirconium terephthalate MOF triggering the electro and photo-chemical reduction of protons to hydrogen (Figure 6).21 Stability and catalytic performance of hydrogenase-like MOF overcome by far the one of the FeFe homogeneous catalyst, demonstrating the benefits of the MOF shell protection of the active catalytic centres.
Oxidative capacity of metalloenzymes can also be applied for a fast detoxification or degradation of persistent pollutants, such as phenols or fluorinated compounds. Bioinspired by this function, J. Wu and co-workers applied a protein engineering inspired approach to boost the oxidase-like activity of several iron-based MOFs through the installation of electron withdrawing functions at the organic pillars of the framework.22 By following this smart strategy, the authors were able to alter the second coordination shell of the metal ions, enhancing in parallel the oxidase-like activity of the MOF framework, which achieved a fast oxidation of phenol like model pollutants. In parallel, the structural characteristics of several zirconium and cerium MOFs resemble phosphor-triesterase-like activity to hydrolyse phospho-organic compounds as pesticides and warfare agents.23
In conclusion, reticular chemistry provides chemically and thermally stable frameworks where metal-biocatalysts´ cores can be replicated and installed. Altogether, reticular metalloenzyme-like systems hold great promise to expand the operational window and diversity of biocatalyst. Nevertheless, it is important to note that up to date only two of the six enzymatic main functions have been replicated at MOF-metalloenzymes: oxidoreductase and hydrolase-like activities. So far, bio-metallocatalysts MOFs exhibiting transferase, lyase, isomerase or ligase activities are unexplored, even though the application of chiral MOFs in asymmetric catalysis and enantiomeric separation holds great promise to expand the potentials of reticular chemistry to mimic enzymatic activities.
The perspective to apply reticular chemistry to mimic enzymatic systems is just in its nascent stage. There is still a room for improvement of the MOF-like metalloenzyme systems in order to finely design not only the metal bio-coordination cores of enzymes, but to fine tune their second coordination shell and surrounding pocket by clusters and linker multivariate encoding. The rational design of asymmetric pores having anisotropic properties (i.e. electric field, hydrophilic/hydrophobic nature…) will be a key milestone to approach the great specificity of enzymes to recognize single substrates. Far from the usual pre or post synthetic functionalization chemistry of MOFs, molecular imprinting could have a great potential to control the specific location and disposition of amino acid residues within pore space of MOFs. In parallel, we can benefit from the dynamic behaviour of flexible MOFs to endow metalloenzyme reticular materials of a self-adaptable framework able to host specific substrates, achieving their activation by the application of external stimuli that distort or strain the porous framework where the active pocket has been replicated.
It will be fascinating to uncover if once stablished the background of reticular metalloenzyme chemistry, the direct evolution approach usually applied to evolve enzymatic systems can also be applicable to ordered metal-organic frameworks. Going a step forward, systems are as effective at transferring the catalytic products within the coupled cascade process, as they are at transforming them. Therefore, if the abovementioned objectives are challenging themselves, to find out whether the biocatalytic activity of different metalloenzyme MOFs can be coupled in a cascade mode will be a fascinating concept to explore beyond the current research frontier.
Acknowledgements
The authors thank financial support from the Spanish Ministry of Economy and Competitiveness (MINECO) through projects MAT2016-76739-R (AEI/FEDER, EU) and MAT2016-76039-C4-3-R (AEI/FEDER, UE) (including FEDER financial support) and from the Basque Government Industry and Education Departments under the ELKARTEK, HAZITEK and PIBA (PIBA-2018-06) programs, respectively. The European Commission Research & Innovation H2020-MSCA-RISE-2017 (Ref.: 778412) INDESMOF project is also acknowledged. Ainara Valverde acknowledges the Basque Government (Education Department) for her PhD grant (PREB_2018_1_004). Leire Celaya-Azcoaga thanks the University of the Basque country for the PhD fellow. The authors thank the technical and human support provided by SGIker (UPV/EHU).
References
(1) Heux, S.; Meynial-Salles, I.; O’Donohue, M. J.; Dumon, C. White Biotechnology: State of the Art Strategies for the Development of Biocatalysts for Biorefining. Biotechnology Advances 2015, 33 (8), 1653–1670.
(2) Commision, E. The European Green Deal. COMMUNICATION FROM THE COMMISSION TO THE EUROPEAN PARLIAMENT, THE EUROPEAN COUNCIL, THE COUNCIL, THE EUROPEAN ECONOMIC AND SOCIAL COMMITTEE AND THE COMMITTEE OF THE REGIONS 2019.
(3) Desage-El Murr, M. Nature Is the Cure: Engineering Natural Redox Cofactors for Biomimetic and Bioinspired Catalysis. ChemCatChem 2020, 12 (1), 53–62.
(4) Zastrow, M. L.; Pecoraro, V. L. Designing Functional Metalloproteins: From Structural to Catalytic Metal Sites. Coordination Chemistry Reviews 2013, 257 (17–18), 2565–2588.
(5) Fontecilla-Camps, J. C.; Amara, P.; Cavazza, C.; Nicolet, Y.; Volbeda, A. Structure-Function Relationships of Anaerobic Gas-Processing Metalloenzymes. Nature 2009, 460 (7257), 814–822.
(6) Moura, I.; Pauleta, S. R.; Moura, J. J. G. Enzymatic Activity Mastered by Altering Metal Coordination Spheres. Journal of Biological Inorganic Chemistry 2008, 13 (8), 1185–1195.
(7) Ji, Z.; Wang, H.; Canossa, S.; Wuttke, S.; Yaghi, O. M. Pore Chemistry of Metal–Organic Frameworks. Advanced Functional Materials 2020, 30 (41), 2000238.
(8) Waldron, K. J.; Rutherford, J. C.; Ford, D.; Robinson, N. J. Metalloproteins and Metal Sensing. Nature 2009, 460 (7257), 823–830.
(9) Thompson, Z.; Cowan, J. A. Artificial Metalloenzymes: Recent Developments and Innovations in Bioinorganic Catalysis. Small 2020, 16 (27), 2000392.
(10) Wei, T. H.; Wu, S. H.; Huang, Y. Da; Lo, W. S.; Williams, B. P.; Chen, S. Y.; Yang, H. C.; Hsu, Y. S.; Lin, Z. Y.; Chen, X. H.; Kuo, P. E.; Chou, L. Y.; Tsung, C. K.; Shieh, F. K. Rapid Mechanochemical Encapsulation of Biocatalysts into Robust Metal–Organic Frameworks. Nature Communications 2019, 10 (1), 1–8.
(11) Davis, H. J.; Ward, T. R. Artificial Metalloenzymes: Challenges and Opportunities. ACS Central Science 2019, 5 (7), 1120–1136.
(12) Freund, R.; Canossa, S.; Cohen, S. M.; Yan, W.; Deng, H.; Guillerm, V.; Eddaoudi, M.; Madden, D. G.; Fairen-Jimenez, D.; Lyu, H.; Macreadie, L. K.; Ji, Z.; Zhang, Y.; Wang, B.; Haase, F.; Wöll, C.; Zaremba, O.; Andreo, J.; Wuttke, S.; Diercks, C. S. 25 Years of Reticular Chemistry. Angewandte Chemie International Edition 2021.
(13) Peng, J.; Wu, R. Metal–Organic Frameworks in Proteomics/Peptidomics-A Review. Analytica Chimica Acta 2018, 1027, 9–21.
(14) Cohen, S. M. Postsynthetic Methods for the Functionalization of Metal-Organic Frameworks. Chemical Reviews 2012, 112 (2), 970–1000.
(15) Serre, C.; Devic, T. Iron and Groups V- and VI-Based MOFs. The Chemistry of Metal–Organic Frameworks: Synthesis, Characterization, and Applications 2016, 171–202.
(16) Sun, Q.; Aguila, B.; Ma, S. Metalloenzyme Mimicry at the Nodes of Metal-Organic Frameworks. Chem 2018, 4 (12), 2736–2738.
(17) Xiao, D. J.; Bloch, E. D.; Mason, J. A.; Queen, W. L.; Hudson, M. R.; Planas, N.; Borycz, J.; Dzubak, A. L.; Verma, P.; Lee, K.; Bonino, F.; Crocellà, V.; Yano, J.; Bordiga, S.; Truhlar, D. G.; Gagliardi, L.; Brown, C. M.; Long, J. R. Oxidation of Ethane to Ethanol by N2 O in a Metal-Organic Framework with Coordinatively Unsaturated Iron(II) Sites. Nature Chemistry 2014, 6 (7), 590–595.
(18) Ikuno, T.; Zheng, J.; Vjunov, A.; Sanchez-Sanchez, M.; Ortuño, M. A.; Pahls, D. R.; Fulton, J. L.; Camaioni, D. M.; Li, Z.; Ray, D.; Mehdi, B. L.; Browning, N. D.; Farha, O. K.; Hupp, J. T.; Cramer, C. J.; Gagliardi, L.; Lercher, J. A. Methane Oxidation to Methanol Catalyzed by Cu-Oxo Clusters Stabilized in NU-1000 Metal-Organic Framework. Journal of the American Chemical Society 2017, 139 (30), 10294–10301.
(19) Zheng, J.; Ye, J.; Ortuño, M. A.; Fulton, J. L.; Gutiérrez, O. Y.; Camaioni, D. M.; Motkuri, R. K.; Li, Z.; Webber, T. E.; Mehdi, B. L.; Browning, N. D.; Penn, R. L.; Farha, O. K.; Hupp, J. T.; Truhlar, D. G.; Cramer, C. J.; Lercher, J. A. Selective Methane Oxidation to Methanol on Cu-Oxo Dimers Stabilized by Zirconia Nodes of an NU-1000 Metal-Organic Framework. Journal of the American Chemical Society 2019, 141 (23), 9292–9304.
(20) Baek, J.; Rungtaweevoranit, B.; Pei, X.; Park, M.; Fakra, S. C.; Liu, Y. S.; Matheu, R.; Alshmimri, S. A.; Alshehri, S.; Trickett, C. A.; Somorjai, G. A.; Yaghi, O. M. Bioinspired Metal-Organic Framework Catalysts for Selective Methane Oxidation to Methanol. Journal of the American Chemical Society 2018, 140 (51), 18208–18216.
(21) Pullen, S.; Fei, H.; Orthaber, A.; Cohen, S. M.; Ott, S. Enhanced Photochemical Hydrogen Production by a Molecular Diiron Catalyst Incorporated into a Metal-Organic Framework. Journal of the American Chemical Society 2013, 135 (45), 16997–17003.
(22) Wu, J.; Wang, Z.; Jin, X.; Zhang, S.; Li, T.; Zhang, Y.; Xing, H.; Yu, Y.; Zhang, H.; Gao, X.; Wei, H. Hammett Relationship in Oxidase-Mimicking Metal–Organic Frameworks Revealed through a Protein-Engineering-Inspired Strategy. Advanced Materials 2021, 33 (3), 2005024.
(23) Gil-San-Millan, R.; López-Maya, E.; Platero-Prats, A. E.; Torres-Pérez, V.; Delgado, P.; Augustyniak, A. W.; Kim, M. K.; Lee, H. W.; Ryu, S. G.; Navarro, J. A. R. Magnesium Exchanged Zirconium Metal-Organic Frameworks with Improved Detoxification Properties of Nerve Agents. Journal of the American Chemical Society 2019, 141 (30), 11801–11805.
Authors
Itziar Oyarzabal1,2, Ainara Valverde1,3, Leire Celaya-Azcoaga1,4 & Roberto Fernández de Luis,1
1BCMaterials, Basque Center for Materials, Applications and Nanostructures, UPV/EHU Science Park, 48940 Leioa, Spain
2IKERBASQUE, Basque Foundation for Science, 48013 Bilbao, Spain
3Department of Physical Chemistry (Macromolecular Chemistry Group (LABQUIMAC). Faculty of Science and Technology. University of the Basque Country (UPV/EHU). Barrio Sarriena s/n 48940 Leioa, Spain, 48013 Bilbao, Spain
4Department of Inorganic Chemistry. Faculty of Science and Technology. University of the Basque Country (UPV/EHU). Barrio Sarriena s/n 48940 Leioa, Spain, 48013 Bilbao, Spain
1 comment
[…] Material berriak nahitaezkoak izango dira ingurumen-kudeaketan: deskarbonizazioan, erregai jasangarrien produkzioan edota konposatu toxikoen inaktibazioan, esaterako. Honelako materialak garatzeko modurik onena natura imitatzea dela erakusten du BCMaterialsek: Mimicking nature to face the green environmental transition […]