Kinetic modelling of catalysis for the Ostwald process
Kinetic modelling of catalysis for the Ostwald process
A catalyst is a substance that increases the rate of a chemical reaction without itself undergoing any permanent chemical change. The study of enzymes has provided us many of the prototypical concepts of catalysis, including the idea that a catalyst lowers the barrier to reaction and thereby accelerates the approach to chemical equilibrium, without altering the position of this equilibrium.
Enzymes are able to bind reagents to the active site, directing them into an energetically stabilized structure that resembles the reaction’s transition state. This catalytic mechanism is the result of millions of years of evolution that produced steric effects, originally described by a “lock and key” model. These steric effects successfully accelerate reactions at the moderate temperatures of living beings.
Enzymes are homogeneous catalysts, as they are in the same liquid phase as the reactants. On the other hand, man-made industrial catalysts using metal surfaces are heterogeneous catalysts as they are in different phases. This is the main reason reactions on metal surfaces often deviate strongly from the lock and key model.
Industrial catalysts typically operate at elevated temperatures enhancing the influence of desorption and there are no lock or key that bring the reactants to the active site. Instead, surface diffusion accomplishes this function; thus, a reactant’s ability to diffuse on the catalyst surface to the active site while competing against thermal desorption may determine the catalyst’s activity. This points out the importance of accurate determination of thermal diffusion coefficients and desorption rate constants under realistic reaction conditions on catalytic surfaces.
The high coverages produced under industrial high-pressure conditions often lead to complex steady-state surface structures involving interactions between different adsorbates. As a result, the potential energy landscape for diffusion of one reactant may depend strongly on the presence (or absence) of the other. Determining this energy landscape and how it depends on surface structure and adsorbate concentration represents a fascinating problem in physical chemistry of great relevance to understanding catalytic behaviour.
Little is known about the impact of co-adsorbate interactions on reactant and transition state energies and entropies. The situation is not made easier by the fact that state-of-the-art electronic structure theory – density functional theory (DFT) – is unable to describe van der Waals interactions that are thought to be crucially important to a correct description of adsorbate−adsorbate interactions. Another approach to the problem was needed.
This is where the introduction of velocity-resolved kinetics (VRK) method makes a difference, as it allows measuring the kinetic competition of diffusion and desorption under industrially relevant high-temperature. This method provides highly accurate desorption rate constants that can be modelled by statistical rate theories to yield adsorbate binding energies and entropies.
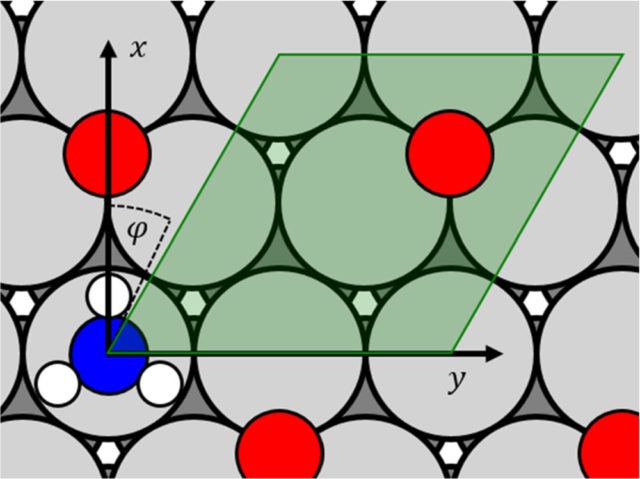
NH3 diffusion on Pt(111) is remarkably slow, due to an unexpectedly high diffusion barrier. This calls into question the wisdom of a generally applied assumption in the modelling of industrial catalysis that diffusion is not a rate-determining process. Within the context of NH3 oxidation on Pt, a team of researchers has now hypothesized 1 that NH3 diffusion could be further influenced by co-adsorbed oxygen atoms on Pt(111), which will be also present at the catalyst under industrial ammonia oxidation conditions.
The team has produced a detailed VRK study of NH3 desorption and diffusion on O/Pt(111) that confirms that there is no evidence for NH3 decomposition or oxidation on Pt(111), which means that the observed differences in desorption rates are attributed to non-reactive interactions between NH3 and O. Using a non-covalent additive interaction model describing attractive and repulsive forces acting between NH3 and O on Pt(111), they could reproduce the observed thermal desorption rates quantitatively over a broad temperature range. In this way, the model explains the steric interactions between adsorbed O-atoms and NH3 molecules.
These results provide critical benchmarks for first-principles calculations of non-reactive interactions between NH3 and O on Pt(111). Comparing these quantities to DFT predictions reveals that dispersion corrections are crucial for obtaining a chemically accurate description of the energetic landscape. These findings set a strong basis for a comprehensive kinetic modelling study of ammonia oxidation for the Ostwald process.
Author: César Tomé López is a science writer and the editor of Mapping Ignorance
Disclaimer: Parts of this article may have been copied verbatim or almost verbatim from the referenced research papers
References
- Dmitriy Borodin, Oihana Galparsoro, Igor Rahinov, Jan Fingerhut, Michael Schwarzer, Stefan Hörandl, Daniel J. Auerbach, Alexander Kandratsenka, Dirk Schwarzer, Theofanis N. Kitsopoulos, and Alec M. Wodtke (2022) Steric Hindrance of NH3 Diffusion on Pt(111) by Co-Adsorbed O-Atoms JACS doi: 10.1021/jacs.2c10458 ↩