Repurposing ponatinib for ALT-specific minority types of recurrent tumors
Repurposing ponatinib for ALT-specific minority types of recurrent tumors
Author: José R. Pineda got his Ph.D. from University of Barcelona in 2006. Since 2007 he has worked for Institut Curie and The French Alternative Energies and Atomic Energy Commission. Currently he is a researcher of the UPV/EHU. He investigates the role of stem cells in physiologic and pathologic conditions.
Replicative senescence is a process that occurs when the nucleoproteic structures of genetic material at the end of chromosomes are progressively lost during cell division. Cancer cells are very active undergoing cell proliferation and contrary to normal cells does not reach replicative senescence. This is possible because cancer cells are able to protect these nucleoproteic structures using different strategies; To re-express a reverse transcriptase called telomerase that add repeated TTAGGG sequences at the chromosome ends, strategy used by the majority of cancer cells or to rely on another different mechanism independent of telomerase known as ALT (from “Alternative Lengthening of Telomeres”) that is based on homology-directed repair mechanisms. This last strategy is the one used by a minority subset of cancer cells including osteosarcoma, soft tissues sarcoma and lethal brain cancer glioblastoma.
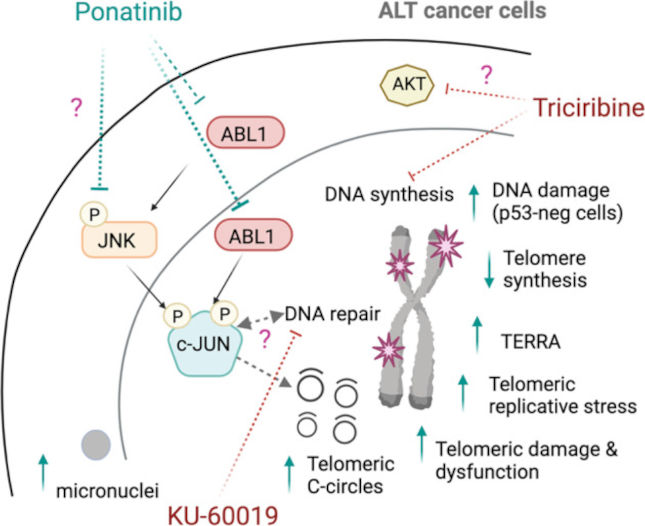
The work of Frances Karla Kusuma and collaborators 1 unravel the characterization and use of ponatinib (a multi-targeted tyrosine-kinase inhibitor) as a chemotherapeutic repurposed medicine to disrupt ALT mechanism. To arrive at this discovery, the research team initially set out to unmask therapeutic vulnerabilities that could be unique to the telomere maintenance processes carried out through ALT. To do this, they screened different libraries of anticancer compounds on two immortalized cell lines, both derived from the same parental cell line (IMR90): the SW26 (ALT-positive) and the SW39 (telomerase positive or isogenic control). They found that SW26 cell line had more sensitivity for PD173074 and dovitinib (FGF inhibitors), YM201636 (PIKfyve inhibitor), nelarabine (inhibitor of DNA synthesis), cediranib, tivozanib and ponatinib (potent inhibitors of vascular endothelial growth factor (VEGF)). Then, they used the proliferative osteosarcoma and the differentiated liposarcoma cell lines to further validate the lethality of the abovementioned compounds. Using clonogenic assays, they found that ponatinib and PD173074 exerted an enhanced killing of ALT cells comparable to telomerase-positive cells. In ALT cells, telomere recombination activity is marked and promoted by ALT-associated PML bodies (APBs). To assess whether these two drugs interfere with ALT mechanisms, they evaluated the levels of extrachromosomal telomeric C-circles (a quantitative measure of extrachromosomal circular telomeric DNA composed of full C-rich strand and notched G-rich complementary strand). They observed that ponatinib was able to increase C-circles at 24 and 72h without observe any effect in telomerase-positive cells or non-cancerous fibroblast cells. The effect of ponatinib on telomeric C-circles was further validated using a (CCCTAA)4-DIG labelled probe (a nonradioactive technology to label and detect nucleic acids) observing to be dependent of intra-nuclear PML bodies (PML bodies are spheres of 0.1–1.0 µm in diameter with matrix-associated domains that recruit an astonishing variety of seemingly unrelated proteins able to regulate many nuclear functions, including DNA replication, transcription, or epigenetic silencing). These results indicated that ponatinib’s effects on ALT cancer cells depended at least partly on the presence of APBs and the ALT activity.
Next question to address was to determine if drug treatments were able to change the number of PML bodies, however either treatment of ponatinib or PD173074 did not showed any change. Then, the authors hypothesized that the drug-induced accumulation of telomeric C-circles may result from damaged telomeres. To test that they determined by western blot and immunostaining the γH2AX histone (a type of histone that phosphorylates when DNA double-strand breaks appears and it is a gold standard to assess global levels of DNA damage). They found that both drugs caused high levels of DNA damage specifically in ALT cells, but not telomerase positive cells. To evaluate whether these drugs caused specific telomere damage, they quantified the levels of telomere dysfunction-induced foci (TIFs) and they found that only ponatinib increased the frequency of TIFs, as marked by the p53 binding protein 53BP1 and telomere staining. They corroborated the results doing a pulse-field gel electrophoresis of embedded cells and running a Southern blot (technique to migrate DNA in a gel and check the size of its fragments) revealing the presence of telomeric DNA in genomic fragments. Furthermore, doing metaphase spreads (a technique frequently used to analyze karyotype) they found an increase of telomere aberrations. Other indicators of potential dysfunction or replication stress at telomeres are the presence of Telomeric RNA molecules (TERRA) at chromosome ends and the increased frequency of micronuclei. Both were found after ponatinib treatment indicating ongoing genomic instability triggering senescence (increase of cell cycle inhibitor p27) in the majority of ALT cell lines but not in telomerase-positive cells.
To assess whether telomere synthesis was affected by ponatinib and to test whether the increase of C-circles was associated with de novo telomere synthesis they used ALT U2OS cell line (due to its longer telomere length). Doing a pulsed-BrdU incorporation assay followed by a telomeric dot blot in Ponatinib treated U2OS cells they found a reduction of newly BrdU-labelled telomeres evidencing its interference with telomeric synthesis and replication. To corroborate this effect they did the telomere synthesis ATSA assay (a test of ALT telomere DNA synthesis in APBs) observing a telomere reduction in both U2OS and SAOS-2 cells after 18–20h of treatment with ponatinib. These results corroborated that ponatinib-induced telomeric C-circle levels were due to an increase of telomere damage and replicative stress concomitant with reduced telomere synthesis.
Next, the authors decided to characterize its effects in vivo through subcutaneous inoculation of CAL72 ALT osteosarcoma cells into immunodeficient mice and posterior treatment by oral gavage with either ponatinib, PD173074 or vehicle (as a control). Interestingly, both drugs were able to reduce the tumor burden in mice without affecting their body weight (a common side effect in current treatments). Residual tumors from mice treated with ponatinib, but not with PD173074 exhibited a marked reduction in the levels of their telomeric C-circles. Using Telomere Restriction Fragment (TRF) analysis, they observed that remaining tumors from mice treated with ponatinib had a slightly shorter average telomere length when compared to tumors from the control group.
Finally, to identify ponatinib’s mode of action on ALT telomeres they did a phospho-RTK array in two different cell extracts after 6 h of drug treatment. They found that only common hit between them was the gene EPHA2 (ephrin receptor subfamily of the protein-tyrosine kinase family involved in cancer development and progression) and JUN (central member of the AP-1 transcriptional family). Using a SILAC-based quantitative phosphoproteome analysis they found to be less abundant the specific phosphopeptides of EPHA2 and JUN in ponatinib-treated cells. Using the Gene Set Enrichment Analysis (GSEA) from RNA-sequencing gene, they found the changes of gene expression after 24 h of drug treatment showing that ponatinib reduced the expression of gene sets related to either DNA repair or replication. Depletion of JUN and ABL1 using CRISPR-Cas9 system led to an increase in telomeric C-circles. The effect of JUN on telomeric C-circles was further confirmed by a rescue experiment with overexpression of gRNA-resistant JUN in SAOS-2 cells lacking endogenous JUN. Restoring JUN in these cells reduced levels of C-circles, confirming a role of JUN in modulating levels of telomeric C-circles. In contrast, knocking-down of EPHA2 had no effect on telomeric C-circles.
In summary, the use of the FDA-approved multi-receptor tyrosine kinase inhibitor Ponatinib in ALT cells is a powerful tool to increase levels of extrachromosomal telomeric C-circles, mediate telomeric dysfunction by limiting telomere synthesis and increasing replicative stress capable of inducing DNA damages in those ALT cells that are deficient for the p53 protein. Although multiple signaling pathways are involved in preserving telomeres of ALT cells, to repurpose ponatinib to use it to deregulate ALT activity concomitant to current chemotherapeutics may represent a new therapeutic option for ALT cancers.
References
- Frances Karla Kusuma, Aishvaryaa Prabhu, Galen Tieo, Syed Moiz Ahmed, Pushkar Dakle, Wai Khang Yong, Elina Pathak, Vikas Madan, Yan Yi Jiang, Wai Leong Tam, Dennis Kappei, Peter Dröge, H Phillip Koeffler, Maya Jeitany (2023) Signalling inhibition by ponatinib disrupts productive alternative lengthening of telomeres (ALT). Nat Commun. doi: 10.1038/s41467-023-37633-3. ↩