Noncanonical β-catenin interactions promote leukemia-initiating activity in early T-ALL
Noncanonical β-catenin interactions promote leukemia-initiating activity in early T-ALL
Author: Marta Irigoyen is a postdoctoral researcher at CIC bioGUNE
T-cell acute lymphoblastic leukemia (T-ALL) is a hematological malignancy that affects both children and adults. Although with current chemotherapy regimens cure is achieved in ~80% of pediatric patients, adults fare more poorly with only 40% 5-year overall survival 1 2.
Restricted cellular subsets with asymmetrically enriched leukemia-initiating cell (LIC) activity have been reported in human and mouse models of T-cell leukemia 345 67, suggesting that more efficient targeting of LICs could lead to dramatic improvements in patient outcomes. Recent studies have demonstrated that LICs are functionally distinct from bulk cells and modulated by distinct molecular signaling pathways 8. Interestingly, high expression levels of β-catenin have been observed in LICs of both human leukemias and mouse models of T-ALL, highlighting the crucial role of β-catenin signaling in T-ALL maintenance and progression. Nonetheless, the mechanisms that regulate β-catenin activity in LICs remain largely unknown. Thus, the work published by Patrizio Panelli and coworkers reports 9 a new signaling pathway mediated by β-catenin in which the interaction of this molecule and the transcription factor FOXO3 promotes LIC activity in cell subsets, mostly enriched in minimal residual disease (MRD), revealing novel potential therapeutic targets for T-ALL treatment.
The role of β-catenin
To address the role of β-catenin in T-ALL, authors first validated that total and active β-catenin are expressed, although at different levels, in leukemic cell lines. Next, they aimed to identify functionally relevant protein interactions of β-catenin in T-ALL and results highlighted the transcription modulators as the main category of enriched β-catenin-interacting proteins. Of interest, within this subgroup they identified non-canonical interactions (when a new feature in an established pathway that does not fit into the established pathway it is called non-canonical) between β-catenin and the transcription factor FOXO3.
β-Catenin signaling plays relevant roles in the maintenance of normal hematopoietic stem cells and LICs 10. To explore the functional cross talk of FOXO3 and β-catenin signaling in established human T-ALLs, authors selected human T-ALL cells with the highest transcriptional levels of both CTNNB1 (the gene that codes for β-catenin)and FOXO3 genes and inserted FOXO and β-catenin fluorescent reporter constructs to track their expression. After cell purification, FOXO and β-catenin containing cells were grafted into secondary recipients, all of which subsequently developed T-ALL. Interestingly, only a small fraction of the totality of leukemia cells exhibited double transduced activity. Of note, deletion of both FOXO3 and β-catenin resulted in a marked decrease in LIC frequency. On the contrary, the constitutive expression of both β-catenin and FOXO3 in leukemia cells increased the LIC frequency. Thus, these findings suggest that both FOXO and β-catenin signaling may reciprocally contribute to LIC activity in T-ALL.
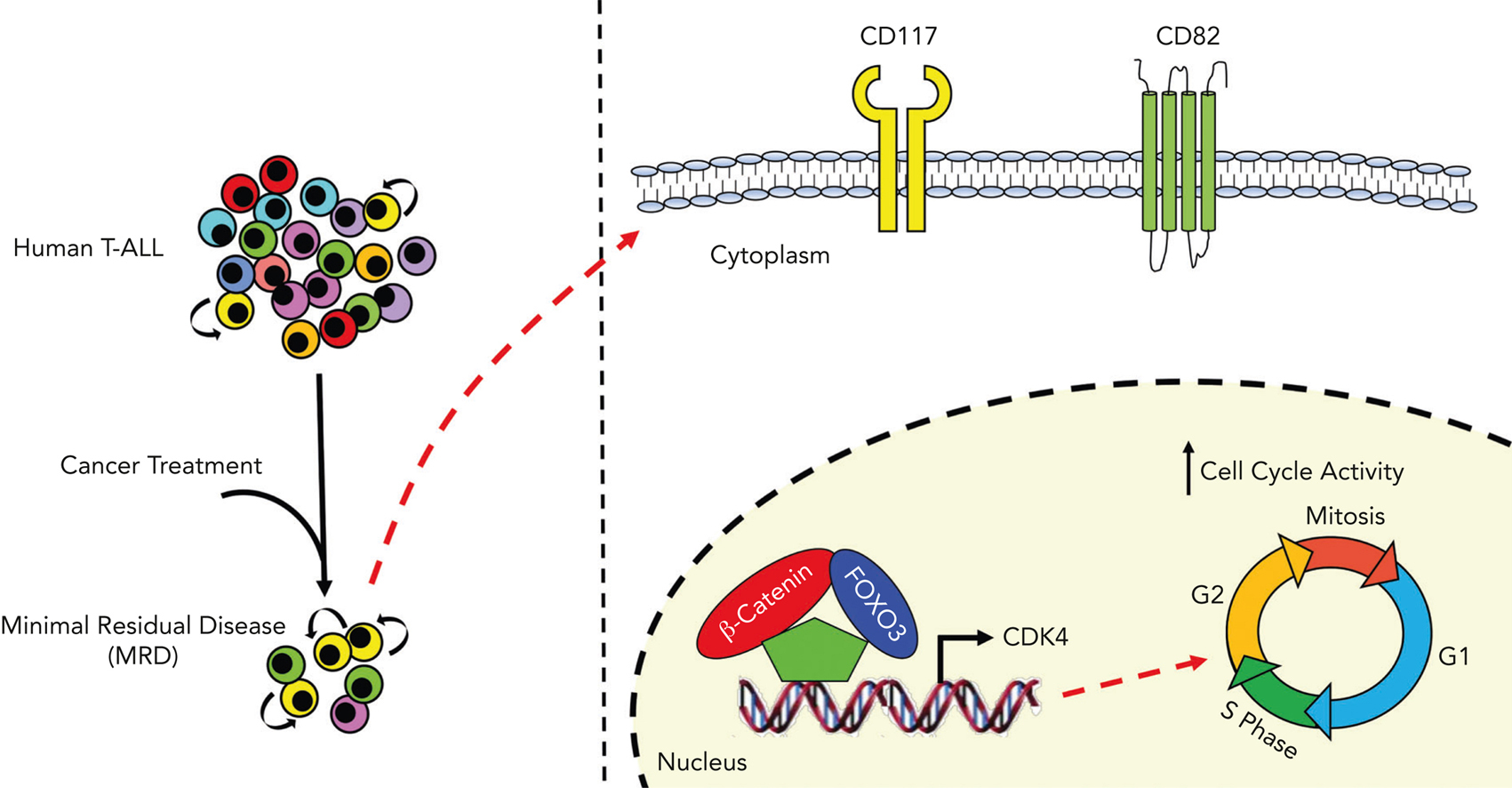
To define the gene expression profiling modulated by either β-catenin or FOXO3 in T-ALL, authors generated double-knockout cell lines to remove FOXO3 and CTNNB1 genes. Afterward, authors introduced FOXO3 and β-catenin genes in FOXO3/CTNNB1 devoided cells to perform RNA-seq assay (a technique used to measure which genes are expressed in a cell sample). Interestingly, gene analysis showed that co-expression of FOXO3 and β-catenin in FOXO3/CTNNB1 devoided cells defined a distinct gene expression profiling. Of interest, the gene set enrichment analysis highlighted the “G2/M checkpoint” as the most enriched hallmark gene set in the gene signature modulated by FOXO3, β-catenin and CDK4. Interestingly, CDK4 is a major modulator of cell cycle during G1/S transition 11 and promotes disease progression in T-ALL 12.
To highlight putative common targets of β-catenin and FOXO3, they generated chromatin immunoprecipitation sequencing data for identifying genome-wide DNA binding sites for β-catenin and FOXO3 in a T-ALL cell line and analyzed them together with publicly available ChIP-seq data sets. They focused their attention on the top gene transcripts and they noted that the genomic region over the transcription start sites of CDK4 gene was enriched in DNA binding sites for both β-catenin and FOXO3, and its expression was significantly induced by β-catenin and FOXO3 in FOXO3/CTNNB1 devoided cells. Moreover, they demonstrated that β-catenin and FOXO3 together induced cycling of FOXO3/CTNNB1 devoided cells. Of note, a similar phenotype was also observed after constitutive expression of a stable isoform of CDK4 (12). Taken together, these data support the conclusion that FOXO3 and β-catenin might drive cell cycling in T-ALL cell lines and that this effect is largely mediated through CDK4 induction.
To define the phenotypic profiling of leukemia cell subsets with high levels of β-catenin and FOXO3, they performed single-cell RNA-seq profiling to detect and quantitatively analyze mRNA molecules in primary samples of human T-ALLs at the time of diagnosis and MRD (Measurable Residual Disease) 30 days after the start of the therapy. After data analysis, they saw that the highlighted cell subset was primarily constituted of MRD cells with a high transcriptional level of β-catenin- and FOXO3-dependent genes, including CDK4. Of interest, they also found that the expression levels of CD82 and CD117 stem cell markers were not modulated by FOXO3 and β-catenin activity, suggesting that these genes are not direct targets of FOXO3 and β-catenin, but surprisingly, LIC activity was enriched in the CD117+/CD82+ fraction. Taken together, these data support the conclusion that CD117 and CD82 may identify LIC-enriched cell subsets, which are considerably heightened in patients with MRD.
To assess the relevance of reported findings in human T-ALL, they evaluated the transcriptional level of both CD117 and CD82 cell markers in a cohort of diagnostic T-ALL samples 13. Strikingly, cases in the highest 25th percentile of transcriptional levels of CD117 and CD82 genes exhibited significantly greater values of MRD compared with other samples, supporting the association between high mRNA expression of CD117 and CD82 and more aggressive clinical disease. Besides, to further characterize the highlighted patients with T-ALL with high CD117 and CD82 mRNA expression at the transcriptional level, authors performed gene set enrichment analysis and they found that patients with high levels of CD117 and CD82 showed a significant enrichment for genes modulated by FOXO3 and β-catenin. These data highlight patients with high CD117 and high CD82 mRNA expression as a distinct subgroup of T-ALL, bearing a gene signature dependent on the activity of both FOXO3 and β-catenin transcription factors.
In conclusion, it would be of high interest to explore the intriguing possibility that therapies that inhibit β-catenin and FOXO3 expression and/or activity may antagonize LIC activity and thereby improve clinical outcomes in patients with T-ALL.
References
- Pui CH, Evans WE. (2006) Treatment of acute lymphoblastic leukemia. N Engl J Med doi: 10.1056/NEJMra052603. ↩
- Giambra V, Jenkins CR, Wang H Lam SH, Shevchuk OS, Nemirovsky O et al. (2012) NOTCH1 promotes T cell leukemia-initiating activity by RUNX-mediated regulation of PKC-theta and reactive oxygen species. Nat Med doi: 10.1038/nm.2960. ↩
- González-García S, Mosquera M, Fuentes P, Palumbo T, Escudero A, Pérez-Martínez A et al. (2019) IL-7R is essential for leukemia-initiating cell activity of T-cell acute lymphoblastic leukemia. Blood doi: 10.1182/blood.2019000982. ↩
- Chiu PPL, Jiang H, Dick JE. (2010) Leukemia-initiating cells in human T-lymphoblastic leukemia exhibit glucocorticoid resistance. Blood doi: 10.1182/blood-2010-06-292300. ↩
- Gerby B, Clappier E, Armstrong F, Deswarte C, Calvo J, Poglio S et al. (2011) Expression of CD34 and CD7 on human T-cell acute lymphoblastic leukemia discriminates functionally heterogeneous cell populations. Leukemia doi: 10.1038/leu.2011.93. ↩
- Tremblay M, Tremblay CS, Herblot S, Aplan PD, Hébert J, Perreault C et al. (2010) Modeling T-cell acute lymphoblastic leukemia induced by the SCL and LMO1 oncogenes. Genes Dev doi: 10.1101/gad.1897910. ↩
- Guo W, Lasky JL, Chang CJ, Mosessian S, Lewis X, Xiao Y et al. (2008) Multi-genetic events collaboratively contribute to Pten-null leukaemia stem-cell formation. Nature doi: 10.1038/nature06933 ↩
- Tamiro F, Weng AP, Giambra V. (2021) Targeting Leukemia-Initiating Cells in Acute Lymphoblastic Leukemia. Cancer Res doi: 10.1158/0008-5472.CAN-20-2571. ↩
- Patrizio Panelli et al (2023) Noncanonical β-catenin interactions promote leukemia-initiating activity in early T-cell acute lymphoblastic leukemia Blood doi: 10.1182/blood.2022017079 ↩
- Giambra V, Jenkins CE, Lam SH, Hoofd C, Belmonte M, Wang X et al. (2015) Leukemia stem cells in T-ALL require active Hif1α and Wnt signaling. Blood doi: 10.1182/blood-2014-10-609370. ↩
- O’Leary B, Finn RS, Turner NC. (2016) Treating cancer with selective CDK4/6 inhibitors Nat Rev Clin Oncol doi: 10.1038/nrclinonc.2016.26. ↩
- Sawai CM, Freund J, Oh P, Ndiaye-Lobry D, Bretz CB, Strikoudis A et al. (2012) Therapeutic targeting of the cyclin D3:CDK4/6 complex in T cell leukemia. Cancer Cell doi: 10.1016/j.ccr.2012.09.016. ↩
- Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR et al. (2017) The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet doi: 10.1038/ng.3909. ↩