Helix-focused peptide libraries for de novo inhibitor discovery
Helix-focused peptide libraries for de novo inhibitor discovery
Protein molecules consist of one or several long chains of aminoacids (usually between and 300 units) called polypeptides – a peptide is an organic compound comprising two or mores aminoacids – linked in a characteristic sequence. This sequence is called the primary structure of the protein. These polypeptides may undergo coiling or pleating, the nature and extent of which is described as the secondary structure.
Protein–protein interactions are essential for cellular function. A common type of interaction found in human cells is the binding of an intrinsically disordered region or a protein (henceforth referred to as IDPs) to a folded domain. Upon binding, the IDP can fold into a distinct secondary structure, often an α-helix. A number of these interactions are involved in disease processes, making these α-helix binding proteins attractive drug targets. However, the large binding interfaces formed by multiple turns of α-helix and a binding protein makes the discovery of inhibitors a significant drug discovery challenge.
One group of compounds showing promise as inhibitors of such interactions is peptides. Peptides can mimic natural IDPs and inhibit otherwise intractable interactions. However, short, linear peptides, much like natural IDPs, tend to be disordered in the absence of a binding partner and often only fold into a distinct secondary structure upon binding. Their folding then comes with an entropic penalty, which negatively impacts the binding strength. To rectify this, macrocyclization – adding a ring of twelve or more atoms – has been proved as a successful strategy to “preorganize” a peptide for binding by stabilizing the structures it adopts upon binding. This has included stabilization of specific secondary structures, and side-chain to side-chain “staples” have been particularly successful in stabilizing α-helices.
A powerful alternative to modifying an existing peptide is screening libraries for de novo macrocyclic peptides. One technology is the RaPID system (random non-standard peptides integrated discovery). The non-standard name refers to the fact that a range of non-canonical amino acids can be included in the peptide library. Affinity selection against an immobilized target protein and subsequent DNA sequencing allows for the identification of de novo cyclic peptide binders. Hit cyclic peptides from RaPID selections form mainly sheet or coil structures, whereas only a few contain α-helix, typically one or two turns. It is not clear how such short N-terminal to side-chain macrocyclic peptides can compete with IDPs that bind using long helices with several turns.
The Bcl-2 (B cell lymphoma 2) protein family contains prominent examples of helical peptides binding to folded partners. For instance, the folded domains of Mcl-1 (myeloid cell leukemia-1) can bind to proteins containing a BH3 (Bcl-2 homologue 3) motif and cause them to fold into a long α-helix. Mcl-1 is an important regulator of apoptosis. By binding to pro-apoptotic proteins containing a BH3 motif, such as Bid (BH3-interacting domain death agonist), Mcl-1 prevents programmed cell death. In cancer, apoptotic mechanisms are often hindered by overexpression of Mcl-1. Higher Mcl-1 expression levels also result in decreased sensitivity to commonly applied anticancer therapies, making Mcl-1 an attractive cancer drug target. Several molecules targeting Mcl-1, including macrocycles, have entered clinical trials, highlighting the importance of Mcl-1 as a drug target.
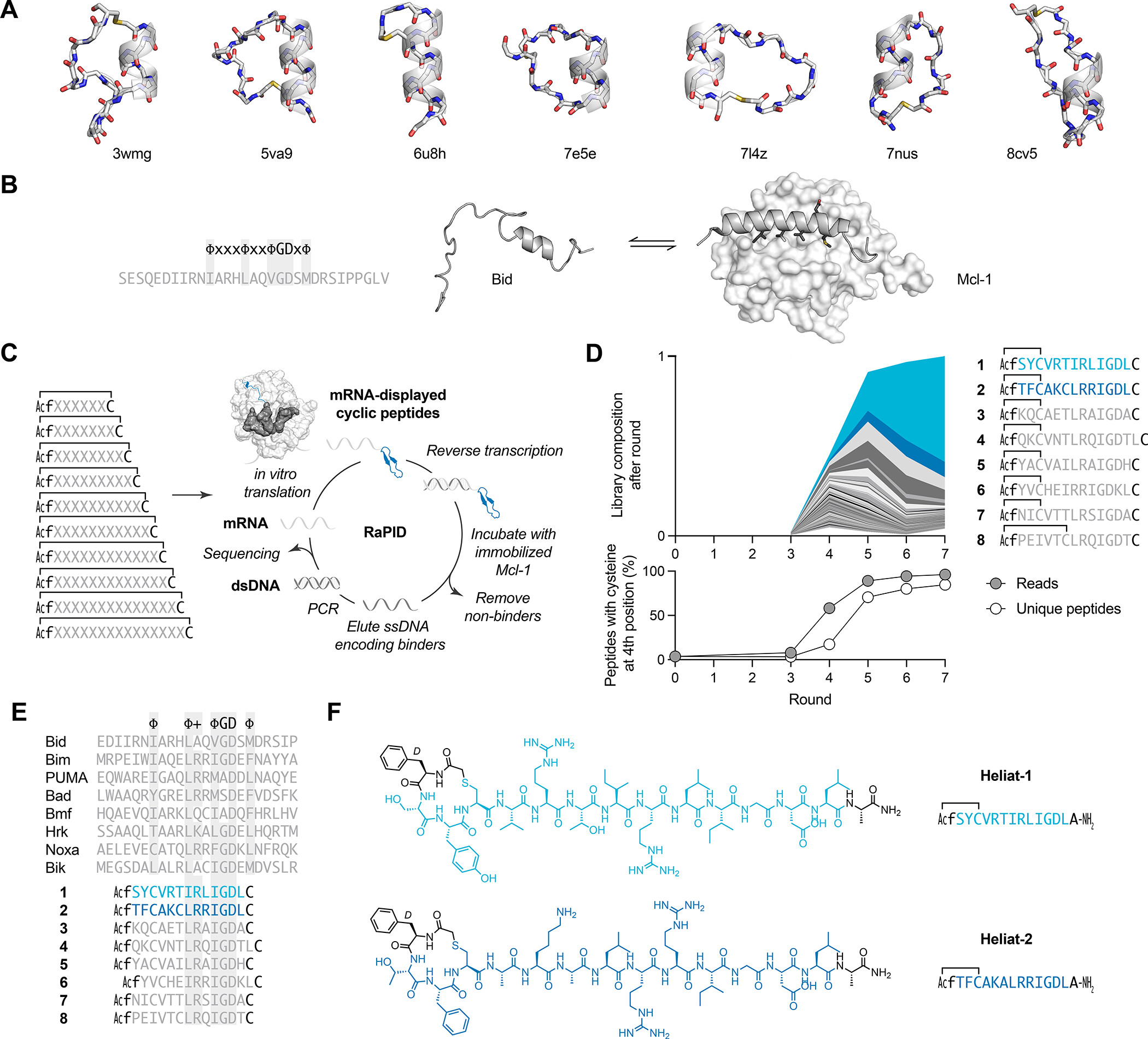
A team of researchers sought to test 1 the ability of RaPID to inhibit the protein–protein interaction between a long helical peptide and a folded protein target, particularly, the ability of N-terminal to side-chain thioether-cyclized peptides to inhibit the α-helix binding protein Mcl-1, by screening a trillion-scale library.
The enriched peptides were lariats featuring a small, four-amino-acid N-terminal macrocycle followed by a short linear sequence that resembled the natural α-helical Mcl-1 ligands. These “Heliats” (helical lariats) bound Mcl-1 with tens of nM affinity, and inhibited the interaction between Mcl-1 and a natural peptide ligand. Macrocyclization was found to stabilize α-helical structures and significantly contribute to affinity and potency.
Yet, the 2nd and 3rd positions within the macrocycle allowed sequence variation, so that a minimal macrocyclic motif could be grafted into a range of peptides and stabilize helical conformations. The researchers found that d-stereochemistry is more helix-stabilizing than l- at the 1st position in the motif. This 15-atom macrocycle, containing a d-stereocenter, could facilitate binding to Mcl-1, improve the inhibitory capacity, and stabilize the α-helical conformations in the discovered peptides.
This mixed stereochemistry macrocyclic N-cap is synthetically accessible, requiring only minor modifications to standard solid-phase peptide synthesis, and its compatibility with peptide screening can provide ready access to helix-focused peptide libraries for de novo inhibitor discovery.
Author: César Tomé López is a science writer and the editor of Mapping Ignorance
Disclaimer: Parts of this article may have been copied verbatim or almost verbatim from the referenced research paper/s.
References
- Fabian Hink, Julen Aduriz-Arrizabalaga, Xabier Lopez, Hiroaki Suga, David De Sancho, Joseph M. Rogers (2024) Mixed Stereochemistry Macrocycle Acts as a Helix-Stabilizing Peptide N-Cap J. Am. Chem. Soc. doi: 10.1021/jacs.4c05378 ↩