How a salt layer shapes electron behaviour in a single molecule
How a salt layer shapes electron behaviour in a single molecule
When scientists place a single molecule on a surface and move electrons in or out one at a time, quantum mechanics comes to life in vivid detail. A team of researchers used a specialized microscope to study a copper-phthalocyanine (CuPc) molecule resting on a thin layer of salt. They discovered 1 that the salt, far from being a passive bystander, actively reshapes the molecule’s structure and decides where extra electrons reside. This finding shows that a molecule’s environment is not just a stage but a key player in its behaviour, opening doors to designing molecule-based electronics or even nanoscale computing systems.
Why study a single molecule?
Electronic engineers typically work with billions of atoms, where individual quirks average out. Chemists, however, focus on the unique properties of single molecules. Scanning tunnelling microscopy (STM) bridges these worlds. By positioning a needle-sharp metal tip within a nanometre of a molecule and applying a voltage, STM makes electrons “tunnel” across the gap, producing a measurable current. This creates images of the molecule’s frontier orbitals—the outermost electron clouds where chemical and electronic activity, like accepting extra electrons, occurs.
To study a molecule’s charge states (neutral, singly charged, or doubly charged), it must be insulated from the metal surface beneath, which would otherwise steal added electrons. The solution is to place a thin layer of sodium chloride (NaCl), just two or three atoms thick, between the molecule and the metal. This salt layer acts like an electronic petri dish, allowing the molecule to hold charges for seconds—long enough for STM to capture detailed images.
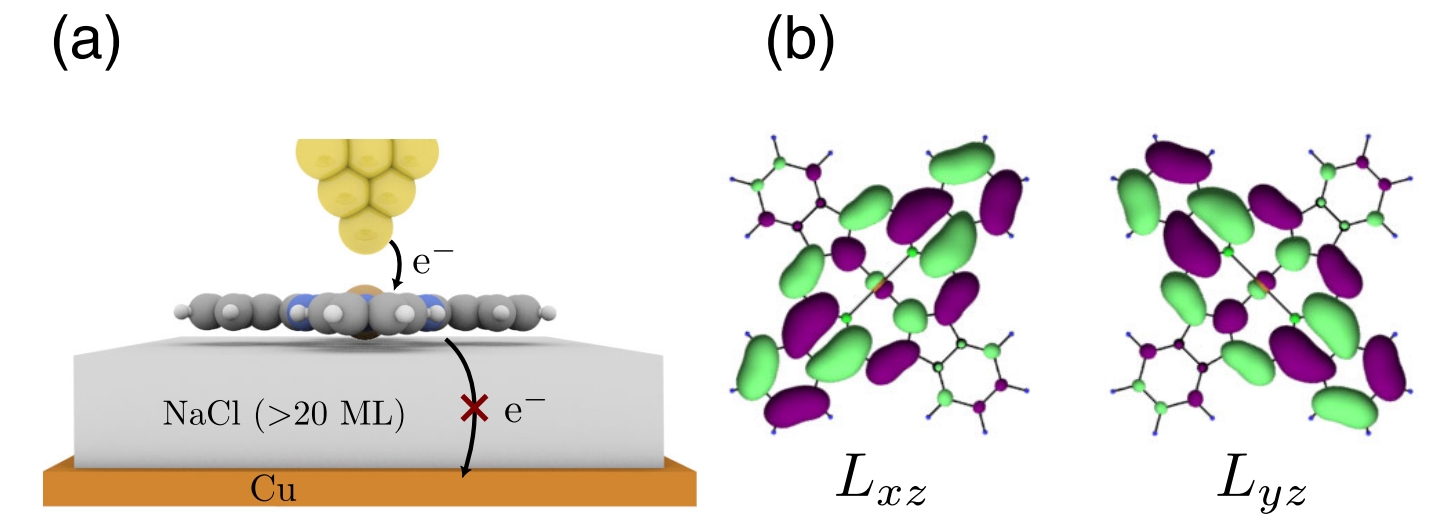
A quantum dilemma
Some molecules, like CuPc, have electron orbitals with identical energies but different shapes, a property called degeneracy. When an extra electron enters such a molecule, it must choose between these equivalent orbitals. Quantum mechanics predicts that the molecule will reshape its atomic framework to favor one orbital, lowering its energy and breaking the symmetry. This reshaping, driven by interactions between the electron and the molecule’s vibrations, is called the Jahn-Teller (JT) effect, first described in the 1930s. For CuPc, which has two nearly identical lowest-unoccupied molecular orbitals (LUMOs), the JT effect kicks in as soon as an electron is added.
Watching electrons move
The researchers used a technique called alternate-charging STM (AC-STM). In AC-STM, the STM tip pulses a voltage to tunnel an electron into the molecule during the positive pulse and remove it during the negative pulse. A tiny quartz cantilever, vibrating like a tuning fork, detects the forces caused by these charge changes with microsecond precision. By scanning the tip across the molecule and measuring how often the electron enters or leaves specific orbitals, AC-STM creates maps showing where the electron is most likely to reside.
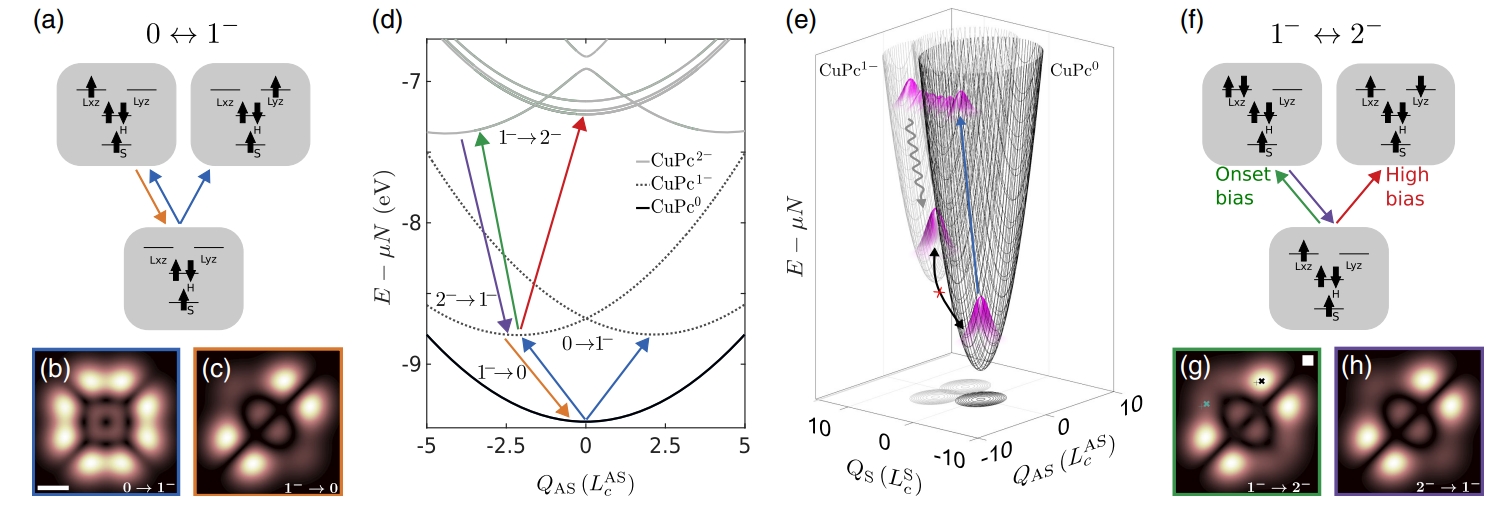
First electron: A classic JT picture
When a neutral CuPc molecule gains one electron (the 0 → 1⁻ transition), the AC-STM map shows a four-fold symmetric pattern, reflecting the equal likelihood of the electron occupying either of the two LUMOs. When the electron is removed (1⁻ → 0), the map collapses to two lobes, indicating that the molecule has distorted to stabilize one orbital (say, Lₓₓ) while disfavoring the other (Lᵧₓ) . This change in symmetry is a hallmark of the JT effect, confirming textbook expectations.
Second electron: An unexpected twist
You might expect a second electron to occupy the empty LUMO to avoid repulsion between the two negatively charged electrons. Surprisingly, the AC-STM maps for the anion-to-dianion transition (1⁻ → 2⁻) look nearly identical to those for the first electron’s departure, showing that both electrons crowd into the same orbital, leaving the other empty. This counterintuitive result, where the electrons pair up despite their repulsion, suggests that something in the system tips the energy balance, overriding the usual Coulomb cost (estimated at about 100 meV) [1].
The salt’s hidden role
The key lies in the NaCl layer. When a charge is added to the molecule, the salt’s positive and negative ions shift slightly, creating an electric field that stabilizes the charge. This process, called polarization, interacts with the molecule’s vibrations in two ways. Symmetric vibrations respond to the total charge, lowering the molecule’s energy whenever an electron is added—an effect similar to a barrier in molecular transistors known as the Franck-Condon blockade. Antisymmetric vibrations, however, react to how the charge is distributed between the LUMOs, amplifying the JT distortion by favouring one orbital over the other.
The team modelled this using a many-body Hamiltonian, which accounts for electron repulsion, electron-vibration interactions, and a small experimental asymmetry. Their calculations revealed two possible configurations for the dianion: a balanced state with one electron in each LUMO, and a polarized state with both electrons in one LUMO. When the salt’s polarization was included, the polarized state became lower in energy by about 100 meV, explaining why the second electron joins the first in the same orbital.
Tuning with voltage
AC-STM’s voltage pulses act like a dial to control the molecule’s charge state. At the lowest voltage where the (1⁻ → 2⁻) transition occurs (2.75 V), only the polarized configuration is accessible. At higher voltages, excited states with one electron in each LUMO become possible, and the AC-STM maps begin to show features of the second orbital, matching the model’s predictions. This ability to switch configurations by adjusting voltage offers a way to manipulate molecular charge patterns.
One charge at a time
Controlling where electrons reside in a molecule has broad implications. It affects how molecules react chemically, emit light, or interact magnetically, and how energy transfers between them. By selecting substrates with different polarization strengths, like lithium fluoride (LiF) or rubidium iodide (RbI), scientists could flip the dianion’s preference, making the electrons spread out instead of pairing up [1]. Such control could enable molecular arrays where charges interact like bits in a computer, paving the way for nanoscale circuits or computing systems based on charge patterns.
This study also challenges gas-phase assumptions, showing that a molecule’s environment can rewrite its electronic behaviour. AC-STM’s microsecond resolution and atomic-scale precision open the door to studying real-time charge dynamics, such as how quickly a molecule relaxes after gaining an electron. Future work could explore how electronic correlations, vibrations, and substrate properties interplay in other molecules, shedding light on phenomena like electron transfer or chemical reactivity at the nanoscale.
The broader takeaway is that nothing in quantum physics exists in isolation. Just as electrons in solids pair with vibrations to become quasiparticles, molecules on surfaces borrow from their surroundings to shape their behaviour. This work vividly shows how a thin salt layer can steer an electron’s path within a single molecule, offering a blueprint for engineering quantum states one charge at a time
Author: César Tomé López is a science writer and the editor of Mapping Ignorance
Disclaimer: Parts of this article may have been copied verbatim or almost verbatim from the referenced research paper/s.
References
- Moritz Frankerl, Laerte L. Patera, Felix Giselbrecht, Thomas Frederiksen, Jascha Repp, and Andrea Donarini (2025) Substrate Polarization Alters the Jahn-Teller Effect in a Single Molecule Phys Rev. Lett. doi: 10.1103/PhysRevLett.134.176203 ↩