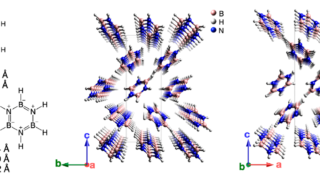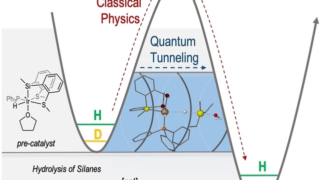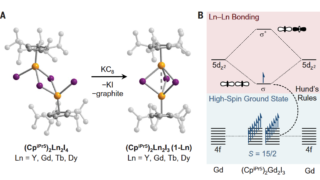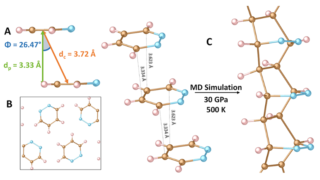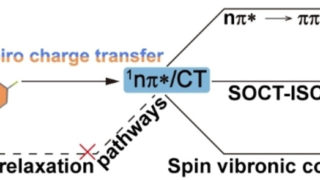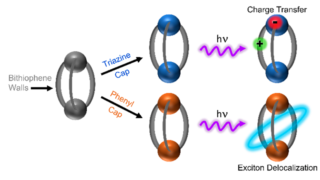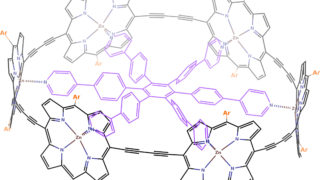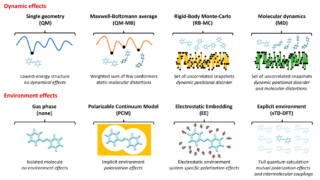
Dynamical nonlinear optical responses of organic materials
Chemistry • DIPC Computational and Theoretical Chemistry • Materials
Nonlinear optics is concerned with the optical properties of matter subjected to intense electromagnetic fields. For nonlinearity to manifest itself, the external field should not be negligible compared to the internal fields of atoms and molecules of which the matter consists. Lasers are capable of generating external fields sufficiently intense for nonlinearity to occur. Actually […]