The Dark Side of Al(III) Chelation Therapy: A New Computational Hope
The Dark Side of Al(III) Chelation Therapy: A New Computational Hope
Author: Gabriele Dalla Torre is a Ph.D. student (ITN-EJD-TCCM) at UPV/EHU
Aluminum is the third most abundant element in Earth’s crust, after oxygen and silicon. As a consequence, during the last century, human intervention has made aluminum so highly bioavailable that C. Exley, one of the leading researcher on Al(III) biochemistry, stated that we are living in the “Aluminum Age”1
Nevertheless, despite its natural abundance, the chemical properties of Al(III) have prevented its presence in the biological cycle of living organisms; moreover, there is increasing evidence that aluminum could be behind of a variety of toxic effects in biological systems, with significant risks for human health.
Indeed, Al(III) was shown to compete effectively with Mg(II) and inhibit Mg(II) dependent enzymatic activities, interfering with the utilization of ATP. It also interferes with enzymatic activities and the secretion of neurotransmitters. Al(III) promotes the hyperphosphorylation of normal neurofilaments, and thus promotes neurofibrillary degeneration. It interacts with β-amyloids, contributing to the formation of amyloid oligomers and then insoluble protein aggregates. These latter processes indicates that the metal ion is neurotoxic, and it has been associated to neurodegenerative diseases, such as the Alzheimer Disease 2
In this rather controversial context, the goal of chelation therapy is the removal of the toxic metal ion from the human body or the attenuation of its toxicity by transforming it into less toxic compounds.
Such a situation led several groups to focus the attention and to make efforts toward the identification of suitable aluminum-specific chelating agents. The main challenges in this sense arise from the fact that all chelators developed so far don’t show specificity only for aluminum, but also for other biologically fundamental cations, like Mg(II), Zn(II) and, above all, Fe(III). This situation clearly leads to a degree of toxicity for these chelating agents, which depends on their affinity toward the metals, condition emphasized by the medical use of EDTA (ethylenediaminetetraacetic acid), one of the most powerful metal chelators3
Moreover, due to the ion’s specific properties, aluminum-containing systems are experimentally complex to be investigated, and complete information on the Al(III)-ligand binding features or the effect of different substituents toward the modulation of the binding affinity are still lacking.
Reflecting on these obscure perspectives, we were wondering:
How can we identify a new, suitable strategy that would beat the Dark Side of Al(III) chelation therapy and thus provide a brighter future for humanity?
We felt the Force, and we got the answer: that’s computational chemistry!
Indeed, in the last two decades, hardware and technological improvements have reached such a level that changed the perspectives of theoretical sciences, allowing very accurate and reliable in silico simulations within the fields of biology, physics and chemistry. This is particularly true for those theoretical methods based on quantum chemistry (such as semiempirical, ab initio and Density Functional Theory ones); nowadays it is possible to perform routinely quantum calculations at medium/high levels of theory with an acceptable computational time, increasing enormously the quality of results and, as a consequence, providing very valuable insights that cannot be always inferred experimentally.
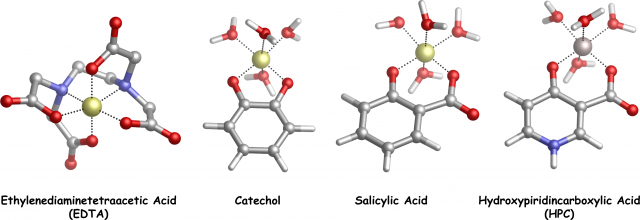
According to that stimulating opportunities, we decided to investigate, by means of state-of-the-art DFT calculations and Bader’s Quantum Theory of Atoms In Molecules analyses, two families of chelating agents (catechols and salicylic acids with different substituents, Fig.1 and Fig.2) which are well known to show high affinity toward Al(III)4
The goal was to set-up a theoretical approach that fits well with the available experimental data, and then to apply it against promising metal chelators, in order to characterize the nature of their interaction with Al(III) and to unveil the effect of different substituents toward the modulation of the binding affinity.
Specifically, the Bader’s Quantum Theory of Atoms In Molecules (QTAIM) calculates the properties of a systems given its wave function; it can be used to predict the nature of the interactions arising within the system, that is, if they are ionic/electrostatic or covalent ones. In our case, we were interested on the nature of the Al-O interactions (Fig.1).
Results that we got from the QTIAM analysis were surprising: although we expected strong ionic interactions between aluminum and the ligands, due to the fact that Al(III) is a plus three charged cation, we found that there is a little degree of covalency in these interactions, present in both families of chelators.
Moreover, and more interestingly, we found that this degree of covalency is modulated by the opposite effect of the substituents; indeed, Electron Donating Groups (EDGs), such as CH3 and OCH3, were found to increase that degree of covalency, while Electron Withdrawing Groups (EWGs), such as NO2, were found to decrease the covalent character of the interactions.
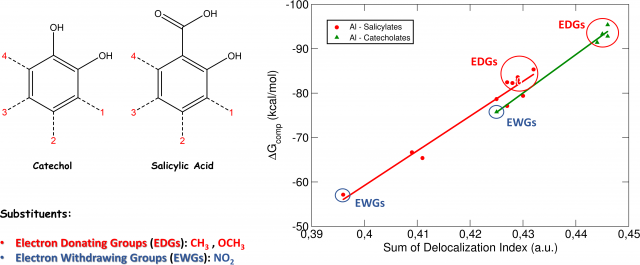
These findings were confirmed by the analysis of the Delocalization Indices (D.I., Fig.2), which measure the average number of electron pairs delocalized (shared) between two atoms.
When Al-O Delocalization Indices are compared with our previously calculated DFT binding energies (ΔG), we can see that there is a clear linear correlation between the two quantities (Fig2): EDGs increase the Delocalization Indices of the complex and, in the same way, increase the binding affinity of the aluminum-chelator system. Conversely, EWGs decrease both the Delocalization Indices and the binding affinity of the complex.
We can interpret these interesting results as follows: Electron Donating Groups, by “pushing” electrons through the aromatic ring, increase the covalent character of the Al-O interaction, which is translated into stronger binding affinities. On the other hand, Electron Withdrawing Groups, by “catching” electrons from the aromatic ring of the molecules, decrease the covalent character of the Al-O interaction, leading to lower binding affinities.
In conclusion, our work allowed us to characterize the nature of the Al-ligand interaction and the role of different substituents on the modulation of the binding affinity. This is a clear example of how state-of-the-art computational approaches allow to get valuable insights that can increase knowledge within those dark points where experimental procedures fail.
Presently, our validated theoretical protocol is going to be applied to other important Al(III) chelating agents, such as EDTA and HPCs (Fig.1).
We feel that we will be able to provide a strong help toward the identification and the tuning of new, potent Al(III) chelators that would defeat once and for all the cruel aluminum’s tyranny.