Autophagy, epilepsy and neurodegeneration: what came first?
Autophagy, epilepsy and neurodegeneration: what came first?
The molecular base underlying neuronal processes is being constantly enriched by extended research focused on the investigation of mechanisms involving neurological pathologies. This broad field ranges from widely extended diseases like Alzheimer’s or Parkinson’s, to other less well-known, generally low-incidence neurodegenerative pathologies. However, one of the most challenging aims of modern neuroscience is the understanding of how these pathways and processes interact with each other; thus, a huge amount of proteins, pathways and concrete mechanisms are being described in the individual context of precise diseases, whereas we still see a very messy and diffuse general landscape of what is really going on inside the human brain. Epilepsy is a pathological process that is present in many different cases of neurological impairment, with as many different clinical manifestations as origins, but the initial triggering events of which remain largely unclear. This is a good example of a common problem in this field of research: the difficulty to assess what came first (the popular what came first the chicken or the egg? still in use although mostly solved by modern evolutionism), and hence, the striking difficulty of addressing the development of an effective medical treatment.
Although the study of neuronal ionic channels has mainly been the first focus of epileptic research, several works in the latest years have found an intriguing link with one of the basic processes that take place in all cells: autophagy. Autophagy was classically simplified as the way in which cells get rid of old structures and unnecessary components; this idea is nowadays deeply enriched with new knowledge, as novel types and regulatory functions for autophagy are being found 1. The role of autophagy in pathologies that evolve with accumulation of intracellular, insoluble materials, is now considered critical and many efforts are made to control autophagy as it is a promising approach.
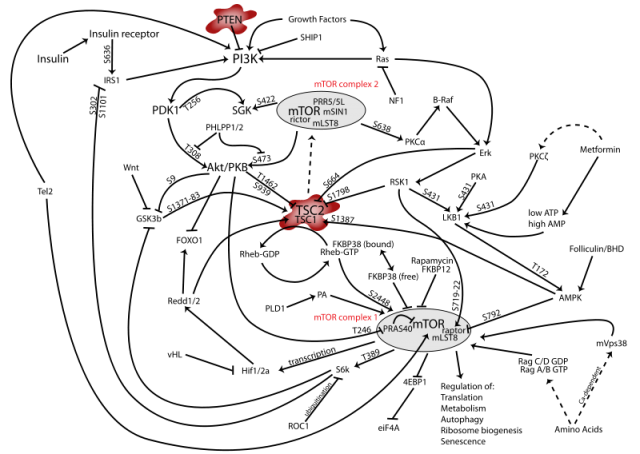
One of the key regulators of autophagy is the pathway controlled by mTOR (mammalian Target of Rapamycin), a protein complex with a central role in different and critical cell signaling pathways (Figure 1); simplifying, activation of mTOR in certain cellular contexts results in autophagy inhibition. A link between this pathway and the development of epileptic seizures had previously been established. In a very recent work published in the Journal of Neuroscience 2, McMahon and collaborators have dealt with the implications of mTOR in epileptogenesis using three different strategies: in a first phase, they have used two different but related types of mice in which the function of a concrete gene is restricted in certain tissues. The selected genes were TSC (Tuberous Sclerosis Complex, an upstream regulator of mTOR) and PTEN (Phosphatase and TENsin homolog, a lipid phosphatase which acts at the first steps of the signaling cascade). This means that the expression of either TSC or PTEN is specifically impeded in neurons from the cortex and the hippocampus of these mice, but not in other regions like the cerebellum. Since both TSC and PTEN are regulators of the mTOR pathway with an inhibitory role, the idea was to disinhibit (or activate, to make it simpler) the signaling cascade and find out if this led to epilepsy. As expected, these mice developed seizures and a molecular analysis of their brain tissue confirmed the increase in the phosphorylated state (meaning activation) of downstream targets of the mTOR complex like the proteins Ulk1 and S6, which are phosphorylated as a result of mTOR activation. This activation was elegantly confirmed to be related to mTOR and not other pathways, since treatment of mice with rapamycin, specific inhibitor of mTOR, produced the opposite effect in the phosphorylation state of both Ulk1 and S6.
It is known that Ulk1 is a critical regulator of the inhibition of autophagy [1], so there was a clear path to follow. To confirm these preliminary results that seemed to suggest a clear link between autophagy and epilepsy in mice models, the authors confirm, in a second phase of the work, the observed effects of mTOR activation using human samples from TSC patients, persons suffering from a disease produced by mutations in the TSC complex (and hence, equivalent to the animal models in which TSC expression was abrogated). Again, these tissues showed a hyperphosphorylation of Ulk1 and S6, together with a marked increase in p62 (another protein that constitutes a typical marker of autophagy), confirming an impairment of the autophagic process.
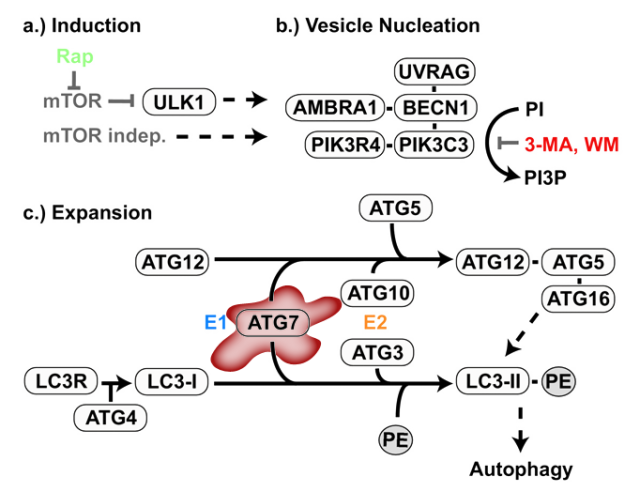
Finally, the third phase was based on a new animal model, a transgenic mouse deficient in another regulator of autophagy: Atg7, a protein involved in the generation of the autophagic structures (Figure 2)3. The purpose of this part of the work was to further confirm the link between autophagy and epilepsy, but avoiding the complex interactive network of mTOR, inhibiting merely the autophagic process from the beginning and trying to observe similar epileptic manifestations. The impairment of autophagy in these mice was confirmed by the increase in p62, among other markers. But the critical question was to observe if this animal model, with an intact mTOR pathway but impaired autophagy, had any relation with epilepsy. Luckily (for the researchers, not for the experimental mice) the animals early showed symptoms of seizures and increased mortality, usually following strong epileptic crises. The message that this final experiment is sending is clear: disruption of autophagy is sufficient for the generation of epilepsy.
The molecular links among neuronal processes like neurodegeneration, impairment of autophagy and development of epilepsy are starting to be elucidated. This could be crucial for several reasons: obviously, the regulation of autophagy is critical in protein clearance pathologies in which accumulation of useless components leads to cell death; but in this work, there is a curious absence of neuronal abnormalities in the mice deficient in Atg7. It is very interesting this separation between epilepsy and neurodegeneration, which may be extremely useful to asses the timing of molecular events in many diseases.
It has been recently described that autophagy is a key process in the development of a rare neurodegenerative disease called Lafora disease 4 . Noteworthy, one of the symptoms of Lafora disease is the apparition of epileptic seizures. Besides, the accumulation of insoluble poliglucosan bodies is a feature that also links this pathology with other similar proteinopathies in which autophagy plays an important role. Putting all these pieces together, the resulting image is a clearer map of how autophagy and accumulation of deleterious material can lead not only to neurodegeneration, but also to development of epilepsy. Addressing the problems with the autophagic pathway can lead to a better clearance of intracellular deposits but also may help to elucidate the origin of epilepsy. Of course one should think that autophagy might just be one of several contributors to epileptogenesis, but likely this knowledge may help to determine the rest of them. If we have the players, and know which of them acts first, it is a matter of time to reach the end of the show.
References
- Singh, R. and Cuervo, A. M. (2011) Autophagy in the cellular energetic balance. Cell metabolism. 13, 495-504 ↩
- McMahon, J., Huang, X., Yang, J., Komatsu, M., Yue, Z., Qian, J., Zhu, X., and Huang, Y. (2012). Impaired autophagy in neurons after disinhibition of Mammalian target of rapamycin and its contribution to epileptogenesis. The Journal of neuroscience: the official journal of the Society for Neuroscience 32, 15704-15714. ↩
- Jaeger, P. A. and Wyss-Coray, T. (2009) All-you-can-eat: autophagy in neurodegeneration and neuroprotection. Molecular neurodegeneration. 4, 16 ↩
- Knecht, E., Aguado, C., Sarkar, S., Korolchuk, V. I., Criado-Garcia, O., Vernia, S., Boya, P., Sanz, P., Rodriguez de Cordoba, S. and Rubinsztein, D. C. (2010) Impaired autophagy in Lafora disease. Autophagy. 6, 991-993 ↩
2 comments
Congrats.
It’s a very interesting article.
I’m very happy with this new ‘mapping’ iniciative. Good luck. 🙂
Thank you pal! We’ll try to do our best to keep the initiative fresh and interesting.
Cheers! ;D