mTOR and rapamycin in autism spectrum disorders
mTOR and rapamycin in autism spectrum disorders
mTOR has nothing to do with Norse mythology, it stands for “mammalian target of rapamycin” or “target of rapamycin in mammalian cells”. mTOR is involved in important cellular processes, including growth, proliferation, motility, survival, protein synthesis, transcription and autophagy.

The history of rapamycin is so suggestive that I cannot avoid mentioning it: in the 1960s, an expedition was made to Easter Island – known by the inhabitants of the island as Rapa Nui – with the aim of identifying natural products from plants and soil with possible therapeutic potential. In 1972, Suren Sehgal identified in these samples a small molecule from the soil bacterium Streptomyces hygroscopicus, which he purified and found to possess potent antifungal activity. He named it rapamycin and subsequent tests revealed that it also had potent immunosuppressive and cytostatic activity against cancer. It is currently used for kidney transplant recipients.
Although ASDs exhibit great genetic and clinical heterogeneity, several ASD syndromes are caused by mutations in genes encoding proteins that inhibit mTOR; that is, these mutations cause loss of inhibition and generate mTOR hyperactivity. Among these genes are Tsc1/Tsc2, NF1 and Pten. mTOR is a key regulator of synaptic protein synthesis, and aberrations in mTOR signaling have been linked to synaptic and neuroanatomical abnormalities seen in syndromic (e.g., tuberous sclerosis) and idiopathic ASDs (1). Among the proteins implicated and that have also been linked to ASD are SHANK3, FMRP and mGluR1/5 glutamate receptors.
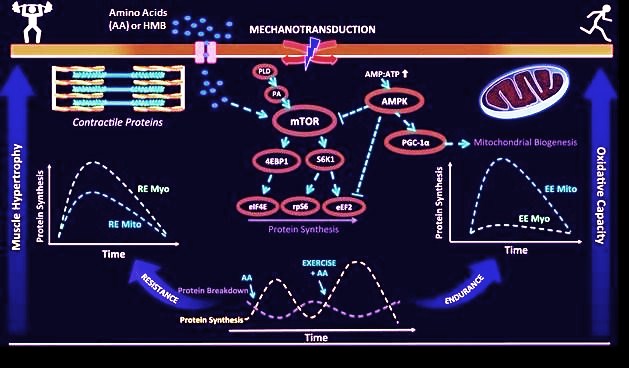
mTOR is involved in the formation of autophagosomes, cytoplasmic vesicles that serve to capture and transport cellular components to lysosomes and there destroy them. This process, known as autophagy or macroautophagy, is important for removing damaged organelles and degrading long-lived or aggregate-prone proteins. Activation of mTOR inhibits autophagy at an early step of autophagosome formation. One theory about the origin of ASD proposes that it is due to an alteration of autophagy by hyperactivation of mTOR. One connection would be that people with ASD have more synapses, because of limited pruning and that pruning is produced by autophagy. Other evidence for a connection would be genetic, since copy number variation mutations have been seen in ASD in genes encoding proteins involved in autophagic pathways. In short, deficits in pruning spines correlate with hyperactivation of mTOR and impaired autophagy. Less pruning implies more spines, and that is what is seen in people with autism.
Further investigations of the mechanisms involved 1, suggest that the observed surplus of excitatory spines could be the result of impaired mTOR-driven autophagy and impaired spine pruning, thus establishing a link between a neuronal signalling abnormality in ASD and the relationship with which postmortem analysis demonstrates synaptic alterations in these disorders.
In Tsc2 +/- mice, an animal model of ASD, and in which mTOR is constitutively hyperactive, Tang et al. (2014) observed defects in postnatal spine pruning, blockade of autophagy, and abnormal social behaviors similar to those in ASD, which fits with everything we have seen above. A striking result is that rapamycin, the mTOR inhibitor, corrected the ASD-like behaviors and synaptic pruning defects in these Tsc2 +/- mice. The results of this article suggest that mTOR-regulated autophagy is required for spine pruning in development, and that activation of neuronal autophagy corrects synaptic pathology and social behavior deficits in mTOR-overactive ASD models.
To determine the presence of synaptic alterations in Tsc2+/- mutants, Pagani et al. 2 measured dendritic spine density in the insular cortex, a region relevant to social dysfunction in autism, and found that Tsc2+/- mice exhibit increased spine density compared to their littermates that served as controls. Next, they used functional MRI to map whole-brain functional connectivity in juvenile Tsc2+/- mutants (P28). The use of prepubertal mice allows the identification of a pattern of connectivity unaffected by the synaptic and circuit remodeling that occurs due to puberty, which as such would be more indicative of the network states that characterize early developmental pathology in ASD.
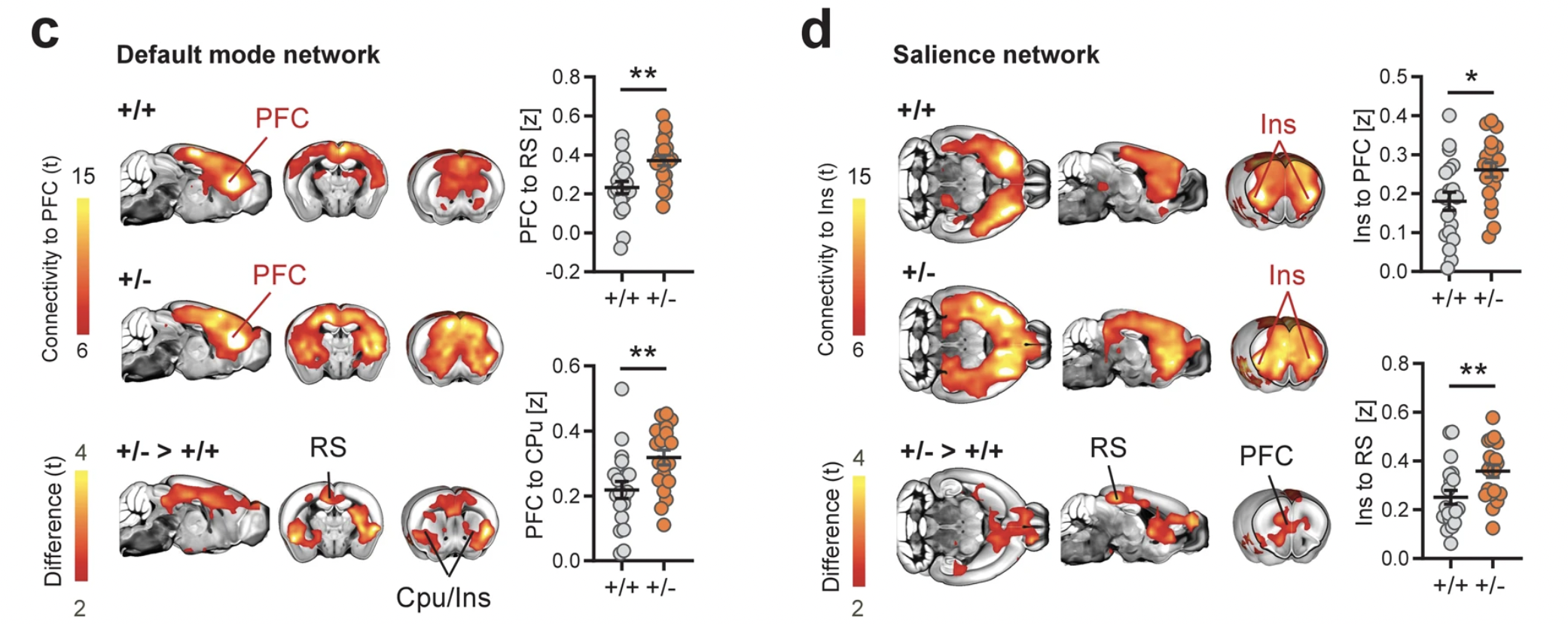
Finally, Pagani et al. treated Tsc2+/- mice and control mice with rapamycin, the mTOR inhibitor, during their fourth postnatal week. Quantification of dendritic spine density revealed that rapamycin treatment rescued spine density in Tsc2+/- mice to return to levels comparable to those of control Tsc2+/+ mice (P < 0.001). Surprisingly, functional MRI of rapamycin-treated Tsc2+/- mice also revealed a complete rescue of the hyperconnectivity phenotype, implying a marked reduction in long-range functional connectivity in the same neocortical and striatal regions characterized by hyperconnectivity in vehicle-treated Tsc2+/- mutants. Consistent with this, rapamycin also rescued default and salience network connectivity in Tsc2+/- mice. These results corroborate the mechanism specificity of our findings and support a causal link between mTOR-dependent synaptic pathology and hyperconnectivity in Tsc2+/- mice.
References
- Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, Sosunov A, Sonders MS, Kanter E, Castagna C, Yamamoto A, Yue Z, Arancio O, Peterson BS, Champagne F, Dwork AJ, Goldman J, Sulzer D (2014) Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83: 1131–1143. ↩
- Pagani M, Barsotti N, Bertero A, Trakoshis S, Ulysse L, Locarno A, Miseviciute I, De Felice A, Canella C, Supekar K, Galbusera A, Menon V, Tonini R, Deco G, Lombardo MV, Pasqualetti M, Gozzi A (2021) mTOR-related synaptic pathology causes autism spectrum disorder-associated functional hyperconnectivity. Nat Commun 12(1): 6084. ↩