Laforin, protein of the year
Laforin, protein of the year
Unveiling the three-dimensional structure of proteins is one of the most useful strategies for understanding the molecular and cellular basis of human pathologies. Since proteins accomplish myriad cellular functions, but they are formed by combinations of only 20 different pieces, the way those pieces are coordinated is critical, and the tiniest difference can change forever the fate of the protein in the complex universe of the intracellular world. Solving the structure of proteins has been a classical scientific challenge for decades, although in the latest years, increasingly growing computing power along with the extended use of synchrotron facilities is making it easier and easier. However, the particularities of each protein are of such diversity that it is quite common to find structures that drive scientists nuts, year after year of struggling for their analysis. This was the case of laforin, a curious protein which shares striking similarities to a particular family of proteins encoded by the human genome, but yet totally different due to some specific traits. The chemical and physical properties of laforin turned the chase for determining its molecular structure into a nightmare for those who dared to pursue this goal (among which I must sadly admit I counted myself). But the good news is that in science, once somebody wins, all of us win; so I am writing this comment with a smile in my face, since after years of imagining and trying to guess the mysteries buried among the 329 amino acids that form the structure of laforin, a central protein in a rare form of epilepsy termed Lafora disease, we can finally behold it.

The winners of such honor have been the researchers led by Matthew Gentry, a long-time collaborator – and at the same time competitor – with whom we have previously been digging into the details of laforin’s structure and biochemical properties. Personally, this trajectory has taught me the huge benefits of scientific collaboration and to what extent a fluid communication is not only good for getting to common goals, but to maintain a healthy and fair competition that has pushed our groups ahead whilst saving us, at the same time, a lot of useless effort. To make a long story short, Gentry’s team had been pursuing laforin’s structure way longer than ours (about 17 years!); but besides, the secret to their success lies largely upon something that is not usually taken into account when talking about human diseases: the clues obtained after thorough study of plant biology and biochemistry. The result of this long and exhausting race were shown this year in Molecular Cell, in an outstanding piece of work with Madushi Raththagala as the first author 1.
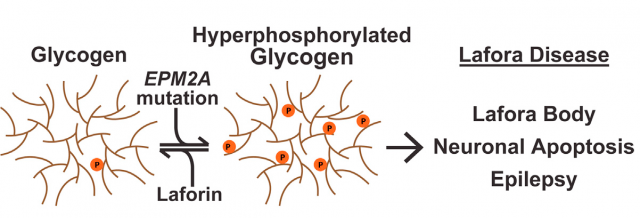
We reviewed the features of Lafora disease, a human pathology in which many different cellular processes are altered, in this post. And although our current research is more headed towards the alterations in autophagy and oxidative stress mediated by laforin and its associated protein malin 23, today we will focus in the central task of laforin as a protein phosphatase with specificity towards glycogen, something unique in the entire human genome. The particular modular composition of laforin, with a highly conserved phosphatase domain (DSP) attached to a carbohydrate-binding module (CBM), seems enough to understand the binding of laforin and glycogen, but biology is far more complex than that. Experimental evidence of direct glycogen dephosphorylation is tricky, but the true oddity in all this business is that at the present time we still do not know the degree of phosphorylation of human glycogen and, what’s more, the origin of the phosphate molecules incorporated into it. For years, Gentry’s lab and others have been approaching this fact from different strategies. Particularly curious is the amount of information gathered in a totally different organism: plants have a molecule similar to our glycogen, starch, with the similar biological function of storing carbohydrates but with some chemical properties that make it different. Most interesting, the aberrant form of glycogen accumulated inside the cells of Lafora disease’s patients is more similar to plant starch than to regular mammalian glycogen. Playing with this similarity, and with the help of plant phosphatases similar to, but evolutionary and functionally unrelated to laforin, finally, the data leading to solving the structure of laforin opens the path to the final understanding not only of the terrible consequences of Lafora disease but to the most accurate comprehension of the biochemical features of glycogen and its metabolism.
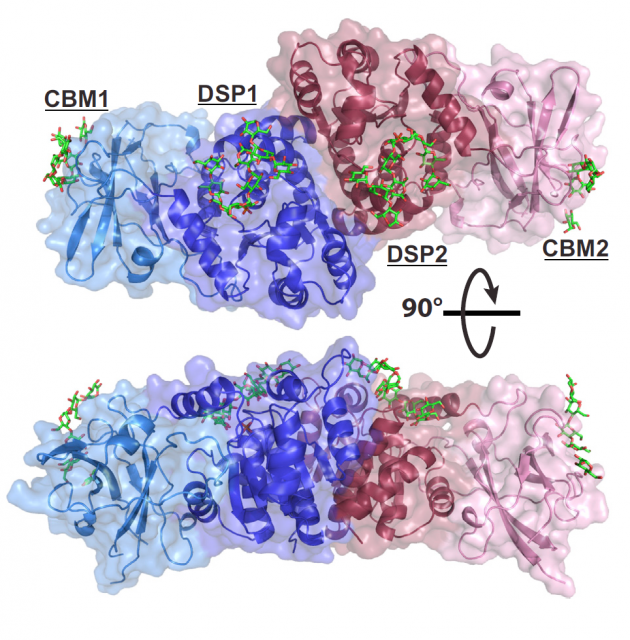
The crystallization of a complete laforin protein bound to maltohexaose (a six-glucose polymer that serves as a model for a piece of glycogen) constitutes a milestone; but the work includes also a nice amount of data obtained from deuterium exchange mass spectrometry (DXMS), dynamic experiments that use the whole protein in solution, and provide complementary information to those obtained with the partial and rigid crystal structure (some amino acids had to be removed to obtain the crystals). Among the most interesting facts revealed by laforin structure is the disposition of both structural domains, which show that glycogen dephosphorylation is strongly dependent on the interface region linking them. This means that mutations in parts of the protein far from the strictly considered carbohydrate-binding domain also affect the capability of laforin to dephosphorylate glycogen. One of the strengths of this work is that the solving of the structure comes along with a fine mapping and directed mutagenesis experiments which provide insightful information on the pathological mutations found in LD patients. Overall, the protein proves to depend on a very tight structural condition to fully accomplish its biochemical role: alterations in either the CBM or DSP parts of the protein affect either the stability of the structure or the binding to glycogen, but in general, they all affect the efficiency of dephosphorylation at some degree. And what’s more, the work finally ends with the controversial issue on laforin dimerization. We and others have previously struggled with this topic and found laforin monomers and dimers in several conditions 4 56, but the details of this modular structure were elusive, until now. The structure is an antiparallel dimer, and destabilization of this arrangement affects binding to the substrate. This modular disposition produces a cooperative effect on the catalytic activity and distinguishes laforin form the arrangements observed in other carbohydrate phosphatases from plants.
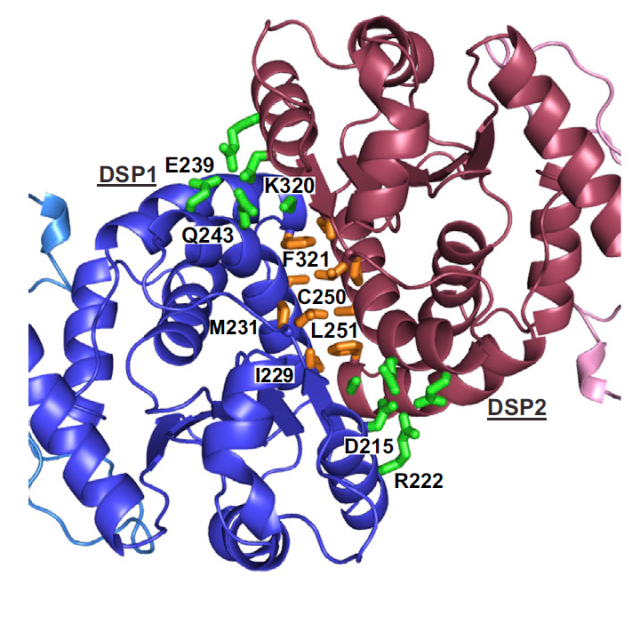
The physiological implications of these findings are of huge relevance: they have permitted to assess that the specificity of the protein is stronger for a particular position of phosphate groups inside the glycogen molecule. Whereas in plants the distribution of phosphate groups added to glucose moieties along the large and branched carbohydrate molecule (starch) is well defined, with the carbon atoms in positions C3 and C6 as the most usual to be phosphorylated, in human glycogen this is still unclear; however, the structure of laforin bound to maltohexaose reveals that the closest atoms to the active site are those in the C3 and C2 locations, being C6 groups further and hence being less probably a target for the protein. Since Lafora bodies have been reported to show an increase in C6-phosphorylated glycogen, with those groups postulated to be normally buried inside the glycogen molecule, it seems that laforin’s activity might be crucial for modulating the accurate branching of glucose molecules by removing C3- and C2-phosphorylated positions and the lack of laforin or the presence of a deficient enzyme then would promote the appearance of C6 phosphates that might bear a more deleterious effect, leading to abnormal glycogen branching, decrease in solubility and, finally, to Lafora bodies formation.
The work was published earlier this year; and a few months ago, I had the pleasure to attend Matthew Gentry’s presentation of these results, having the opportunity to discuss personally the interesting consequences of these findings after years of tough research. But the most exciting fact is that this is not the end of a road: although we finally get to see laforin in its entire splendor, this knowledge means just the beginning of a new series of paths that emerge, and constitute the proof that basic research, biomedicine, and plant biology are not independent branches of knowledge. They fuse together and give us a nice example: digging into a rare, incredibly low prevalence disease, with the help of data obtained from the distant plants – usually underrated in terms of research impact – reveals basic fundamentals of our own biology. The data that laforin structure unfolds are not only relevant to those related to Lafora disease, but to all human beings. This is the kind of work that adds a couple of lines to the still unfinished book of life sciences. In my humble opinion, it truly earns laforin the title of protein of the year.
References
- Raththagala, M., Brewer, M.K., Parker, M.W., Sherwood, A.R., Wong, B.K., Hsu, S., Bridges, T.M., Paasch, B.C., Hellman, L.M., Husodo, S., et al. (2015). Structural mechanism of laforin function in glycogen dephosphorylation and lafora disease. Mol Cell 57, 261-272. DOI: 10.1016/j.molcel.2014.11.020 ↩
- Sánchez-Martín, P., Romá-Mateo, C., Viana, R., and Sanz, P. (2015). Ubiquitin conjugating enzyme E2-N and sequestosome-1 (p62) are components of the ubiquitination process mediated by the malin-laforin E3-ubiquitin ligase complex. Int J Biochem Cell Biol 69, 204-214. DOI: 10.1016/j.biocel.2015.10.030 ↩
- Romá-Mateo, C., Aguado, C., García-Giménez, J.L., Knecht, E., Sanz, P., and Pallardo, F.V. (2015). Oxidative stress, a new hallmark in the pathophysiology of Lafora progressive myoclonus epilepsy. Free radical biology & medicine. DOI: 10.1016/j.freeradbiomed.2015.01.034 ↩
- Sánchez-Martín, P., Raththagala, M., Bridges, T.M., Husodo, S., Gentry, M.S., Sanz, P., and Romá-Mateo, C. (2013). Dimerization of the glucan phosphatase laforin requires the participation of cysteine 329. PLoS One 8, e69523. ↩
- Dukhande, V.V., Rogers, D.M., Romá-Mateo, C., Donderis, J., Marina, A., Taylor, A.O., Sanz, P., and Gentry, M.S. (2011). Laforin, a dual specificity phosphatase involved in Lafora disease, is present mainly as monomeric form with full phosphatase activity. PLoS One 6, e24040. ↩
- Liu, Y., Wang, Y., Wu, C., and Zheng, P. (2006). Dimerization of Laforin is required for its optimal phosphatase activity, regulation of GSK3beta phosphorylation, and Wnt signaling. J Biol Chem 281, 34768-34774. ↩
2 comments
[…] Posted in Noticias, Science, Biochemistry, Molecular biology | 0 comments […]
[…] Conocer la estructura tridimensional de una proteína es fundamental para poder conocer cómo actúa una proteína. Si se trata de una proteína implicada en la aparición o evolución de una enfermedad ese conocimiento es crítico para poder elaborar tratamientos […]