Putting the “rule of five” of drug research in context
Putting the “rule of five” of drug research in context
Lipinski’s “rule of five”,1 very well known by medicinal chemists, has been defined as a set of rules, based on the number 5 -hence its name-, to evaluate the “drug-likeness” of a given molecule. The rules are:
– The molecule should have no more than 5 H-bond donors and 10 H-bond acceptors (i.e. N or O atoms).
– The molecular weight should be under 500 Daltons.
– Its logP should be 5 maximum.

Except that Lipinski’s rule is probably not a “rule” but more of an observation, and it does not apply to all drugs either. Rather, it comes from a data set of known orally administered drugs from which the authors observed that most of them were relatively small and moderately lipophilic. Lipinski and his coworkers thought that it would be useful to use those characteristics to determine if a chemical could have the potential to be orally active in humans, and that gave origin to the popular article that all medicinal chemists have been reading and citing since 1997.
However, a problem of the “rule of five” is, it has been taken so seriously by some that they even could feel “nausea, dizziness and disbelief” when they come across molecules outside of its limits, according to what the medicinal chemist Dave Selwood says in his article about macrocyclic drugs whose molecular weights above 700 obviously do not comply to the rules.2 Meanwhile, others have dismissed the rule’s validity by arguing that many drugs widely used do not follow it at all. Indeed, only a little more than a half of all drugs approved by the FDA comply with the ‘rule of five’. 3 Does this mean the rule is not valid? Not exactly, as it should be put in context, which is something we often miss to do.
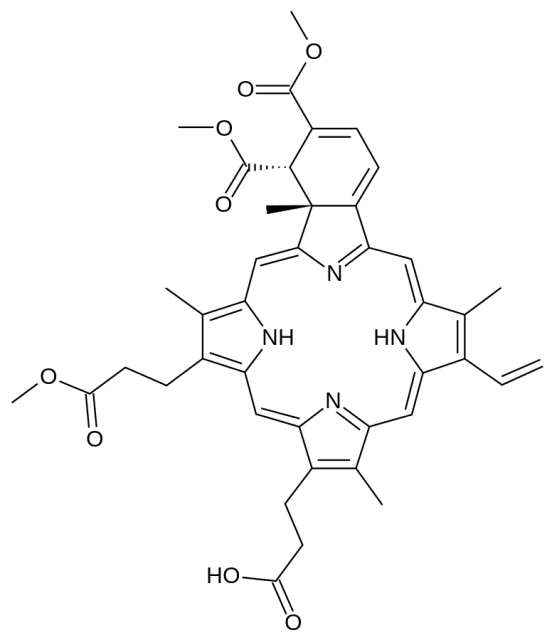
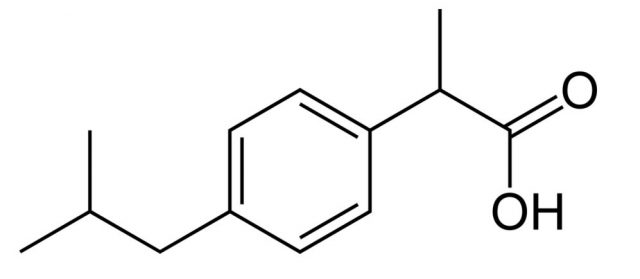
First, this rule is limited because it refers to oral drugs only and it also excludes natural products. This is already excluding a lot of drugs like many broad-spectrum antibiotics and drugs that need to be injected. Like Lipinski himself said in his paper, “the set of rules deals only with solubility and permeability as barriers to drug absorption, while other factors like metabolic events that influence the measurement of drug bioavailability are beyond its scope”. The guidelines were proposed as a response to the huge numbers of random chemical compounds that were being synthesized and screened in the 1990s, partly because of how easy it turned to make entire libraries in a short time when combinatorial chemistry became a thing, and partly because of the search for potency without taking into account other considerations like ADME (absorption, distribution, metabolism, and elimination). Obviously, the potency of a medicine is very important, but if a molecule cannot be correctly absorbed or removed from the body, then its potency is useless. So in an era when everyone started synthesizing and screening huge chemical libraries like there was no tomorrow, Lipinski’s aim was to rationalise compound design and narrow the margins a bit. The basic idea behind his article was: why spend time and effort making very polar and large molecules that, when tested, will have a lower chance of making it to the market as oral drugs when there is plenty of molecules with the “right” characteristics yet to be synthesized and tested?
As an analogy, the phrase “most cars sold are red or black” is a very different statement to “factories should make red or black cars only”. Nevertheless, it gives an idea of what can be successful in the market and it can help manufacturers simplify a car-making process for which there could be too much choice. Nobody is telling manufacturers to not make cars in seven different shades of green and eight of yellow, but not making them won’t affect sales too much and could save a lot of working time and money. Likewise, it surely will be faster and less costly to find orally available drugs if we start by looking at those having the characteristics Lipinksi observed in most compounds already in the market than if we have to go through all the possible molecules synthetic chemists can make. Besides, from a synthetic point of view, the chemical compounds within the boundaries generally coincide with those that can be reasonably handled in the lab during synthesis, work-up and purification. In summary, no one can force you to follow the rule, but using it might spare you a lot of trouble derived from too much choice at the start of a drug discovery project for which, in principle, every molecule, could have an activity and would, therefore, be worth testing.
According to Ming-Qiang Zhang and Barrie Wilkinson (2007), the main problem with taking this rule too seriously is the discovery of oral drugs becoming the aim of too many drug discovery projects: sometimes, oral drugs take too much research effort and resources compared to parenteral (injected) drugs. Besides, it should be considered that perhaps the answer to a problem does not have to necessarily be a small molecule at all, and more research should be done on other alternatives like therapeutic antibodies. In addition, we should not forget about research on natural products that have provided us with very useful therapies throughout history.
References:
1 Lipinski, C. A., et al. “Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings” Adv. Drug Deliver Rev. 1997, 23, 3
2 Selwood DL. Macrocycles, the edge of drug-likeness chemical space or Goldilocks zone? Chem Biol Drug Des. 2017;89:164– 168. doi:10.1111/cbdd.12922.
3 Ming-Qiang Zhang and Barrie Wilkinson. “Drug discovery beyond the ‘rule-of-five’ “. Curr. Opinion Biotechnol. 2007, 18: 478
1 comment
[…] Botika berrien ikerketan bere horretan aplikatzen ez diren arauak badaude. Isabel Pérez Castroren Putting the “rule of five” of drug research in context […]