Antibody-drug conjugates, a fine example of teamwork
Antibody-drug conjugates, a fine example of teamwork
The immune system is a complex network that recognises foreign substances in the body. When it detects an alien substance (antigen), it creates antibodies to attack and destroy it. This system protects vertebrates against viruses, bacteria, fungi and other parasites, and is the reason for the “rejection” after organ transplants. When we have cancer, our bodies also recognise the tumours as foreign and therefore create antibodies to try to attack them, although the immune system is not very successful in this instance -that is why cancer cannot be cured without medicines, since our immune system is insufficient to fight cancer. However, antibodies have a very interesting feature scientists can take advantage of: their specificity. Each antigen has a unique “fingerprint” and a unique antibody that recognises it, and some of those antigens are proteins expressed by cancer cells.
On the other hand, the problem with most available chemotherapies is pretty much the opposite: they are not as specific as we would like them to be due to their strong secondary effects. This is why many patients experience hair loss within their first few chemo sessions: one of the characteristics of cancer cells is that they divide very fast, and many drugs have been designed to halt that fast growth; however, this strategy will not only affect the tumour but also other cells that reproduce at a fast rate such as hair follicles.
The ideal cancer therapy should be one that goes straight to the tumour without affecting other organs, and that is effective against cancer cells. So, in theory, it seems a good idea to combine both elements described above and make them work together: a potent drug that has proved to do its job very well despite not attacking the cancer only, and an antibody that “knows” where to go. The antibody can’t do much against the cancer, but the fact of being so specific would make it able to take the drug where it is required and not anywhere else, thus avoiding unwanted side effects.
In practice, however, this is not that straightforward: once potent chemotherapies have been discovered, it is necessary to identify antigens present on cancer cells either exclusively or at least expressed at a much higher rate than in the rest of the body, otherwise the antibodies will go to the wrong place. And once the antibody and the drug have been found, there is still much chemistry research to do on how to join them together in such a way that the chemical linker is strong enough for the drug not to be released before it gets to the tumour (for example, in the bloodstream where it would cause toxic effects), still easy to break once it has arrived to the cancer cells in order to be able to do its job.
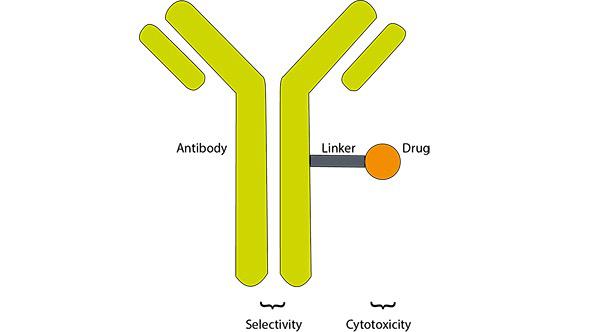
This idea is the basis of antibody-drug conjugates (ADCs) and when it was put in practice for the first time in 1958, it showed to be quite effective when tested against leukemia in hamsters,1 which encouraged researchers to carry on working on it.
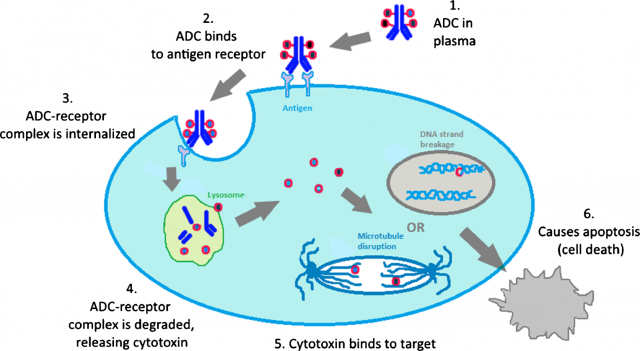
ADCs generally target antigens that are overexpressed at the surface of cancer cells. Once there, they are internalized and the link between the antibody and the drug is cleaved within the cell, releasing the drug in its free form and leaving it ready to work. Examples of this are the two ADCs approved by the FDA so far: Adcetris™ for the treatment of certain hematological malignancies and Kadcyla™ for the treatment of metastatic breast cancer.
Other ADC products currently under preclinical studies have very encouraging results, but this mechanism based in internalization is often associated with drug resistance and poor tumour penetration in solid and/or metastatic tumours. For this reason, much research is done recently in alternative non-internalizing ADCs.2
Alberto Dal Corso and his colleagues have been studying this non-internalization possibility and recently published a paper in which they show how they developed a chemical linker that is stable in serum but can be efficiently cleaved in the extracellular matrix by enzymes released by the dying tumour cells, rather than inside them, and then attack cancer cells.3
Non-internalizing ADCs generally carry tubulin-targeting (antimitotic) drugs: by disrupting tubulin, the microtubules that play an essential role in mitosis cannot function normally, and the cells cannot reproduce. But lately, there has been some research in ADCs incorporating drugs whose mechanism of action is the disruption of the DNA synthesis. This is what the drug used for Dal Corso’s studies, PNU159682, does. When it is free (not linked to an antibody), this drug is active against the four cell lines tested in this study: epidermoid carcinoma, metastatic breast cancer, glioblastoma, and renal cancer.
First of all, a derivative of PNU159682 was chemically linked to a non-internalizing antibody specific to tenascin, one of the most abundant proteins expressed in solid tumours,4 5 lymphomas,6 and in the bone marrow of acute leukemias,7 while being undetectable in normal tissues.8 The ADC developed by these researchers has the structure shown below:
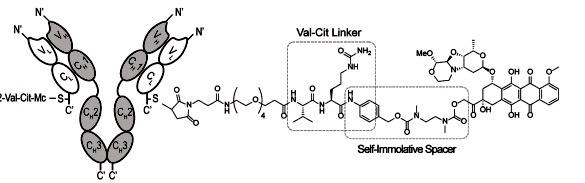
On the left-hand side, with the Y shape, is the antibody which is linked to a chemical chain through a sulfur atom from a cysteine residue (cysteine is an aminoacid that has SH groups). The chain between the antibody and the drug incorporates a polyethylene glycol (PEG) fragment, that is generally added in order to improve the solubility of the complex, as these compounds tend to be insoluble but PEG is soluble in water. In this case, a PEG-4 has been chosen.
The valine-citrulline linker was incorporated to the ADC because these two aminoacids, when found together, are recognised by proteases and, when the peptide bond between them is cleaved, this triggers a “chain” mechanism by which the self-immolative spacer breaks too and releases the drug (the structure with seven rings that appears on the right).
The proteases that trigger this process are mainly intracellular, but they are also released in the extracellular space upon tumour cell death. The acidic tumour microenvironment triggers drug release by proteolysis.9 Also, it is important to notice that, in ADCs, each antibody is conjugated to more than one molecule of the drug. In this case, each antibody was conjugated to two molecules of the drug.
First, the stability of the linker was tested in mouse serum in an incubator, to see how would the ADC behave in a biological medium and to test if the linker would be strong and stable enough whilst travelling through the body on its way to the tumour. After 120 hours, no release of the drug was observed, and all of it showed to still be conjugated to the antibody. This proved the ADC successful to be tested in the next stage on several cancer cell cultures such as kidney cancer, glioblastoma and breast cancer, which proved to be killing the cells.
The ADC exhibited a potent anti-tumour effect in vivo: administration to live mice in which epidermoid carcinoma had been implanted, followed by ex vivo immunofluorescence detection, revealed that the ADC efficiently localized around the tumours. When administered two injections of 1mg/kg of ADC per kg of body weight (mg/kg bw) tumour sizes decreased substantially and the tumour growth stopped for twenty days. When the dose was doubled (injections of 2mg/kg bw), three out of five mice were completely cured but there was toxicity manifested as weight loss. These assays showed that the chemical linker between the antibody and the drug was degraded in the tumour surroundings, that the drug actually worked, and also allowed to find a preliminary dose that was not toxic.
This study confirms the hypothesis that non-internalizing ADCs can work efficiently, that internalization is not a strict requirement for antibody-based therapies, and that there may be more applications of non-internalizing ADCs for cancer therapy than previously assumed. Extracellular matrix proteins are abundant and easily accessible in extracellular areas of most solid tumours, lymphomas and leukemias, and they represent promising targets for antibody-based therapies.
References:
1 Mathé, G, et al. “Effet sur la leucemie 1210 de la souris d’un combinaison par diazotation d’A-methopterin et de γ-globulines de hamsters porteurs de cette leucemie par heterograffe”. C. R. Acad. Sci. 1958, 216:1626–1628.
2Joubert, N et al. “Towards antibody-drug conjugates and prodrug strategies with extracellular stimuli-responsive drug delivery in the tumour microenvironment for cancer therapy”. Eur. J. Med. Chem. 2017, article in press. https://doi.org/10.1016/j.ejmech.2017.08.049
3Dal Corso, A., et al. “A non-internalizing antibody-drug conjugate based on an anthracycline payload displays potent therapeutic activity in vivo”. J. Contr. Release 2017, 264, 211. https://doi.org/10.1016/j.jconrel.2017.08.040
4Dueck, M., et al. “Detection of tenascin-C isoforms in colorectal mucosa, ulcerative colitis, carci-nomas and liver metastases”. Int. J. Cancer, 82 (1999), 477
5Adams, M., et al. “Changes in tenascin-C isoform expression in invasive and preinvasive breast disease”. Cancer Res., 62 (2002), pp. 3289-3297
6Schliemann, C., et al. “Three clinical-stage tumour targeting antibodies reveal differential expression of oncofetal fibronectin and tenascin-C isoforms in human lymphoma”. Leuk. Res., 33 (2009), pp. 1718
7Gutbrodt, K. L., et al. “Antibody-based delivery of interleukin-2 to neovasculature has potent activity against acute myeloid leukemia”. Sci. Transl. Med., 5 (2013), p. 201ra118
8Castellucci, M., et al. “The human placenta: a model for tenascin expression”. Histochemistry, 95 (1991), pp. 449-458
9 Choi, K.Y. et al. “Protease-activated drug development”. Theranostics 2 (2012) 156.
3 comments
[…] Antigorputzak bezain espezifikoa eta farmakorik potenteena bezain eraginkorra dena da minbiziaren aurkako terpia ideala. Zelan ez zaio inori buratu? Agian bai, Isabel Pérez Castro hau bearu lortzeko dabil lanean Antibody-drug conjugates, a fine example of teamwork […]
[…] La terapia ideal contra el cáncer es una que sea tan específica como los anticuerpos y tan efectiva como el fármaco más potente. ¿Cómo no se le había ocurrido esto a alguien? O sí, Isabel Pérez Castro trabaja precisamente […]
I find this forum very interesting. There’s a lot of useful information here.