Exploiting DNA packaging
Exploiting DNA packaging
The haploid human genome contains approximately 3,000,000,000 base pairs of DNA packaged into 23 chromosomes. Most cells in the body, except for eggs and sperm, are diploid, with 23 pairs of chromosomes. That makes a total of 6,000,000,000 base pairs of DNA per cell that, if they were to be put in a straight line, would have approximately 1.8 m length.1 DNA compaction is such that the entire genome of a single human cell is squeezed into a cell nucleus with an average diameter of 6 μm. In other words, the circumference of an average mammalian cell nucleus is almost one million times smaller than the length of the genome that needs to be packed into it.2 This means that DNA must be very tightly compacted to fit into the cell, and it implies the need for a high degree of structural organization: it is not enough to just fold the DNA into a very small space, but the packaging must permit access to the information stored in it without affecting or changing the DNA sequence and properties, and this is achieved thanks to the role of supercoiling and its regulation. This is an important and highly regulated aspect of DNA tertiary structure that is found in every cellular DNA, whose study has provided many insights into DNA’s structure and function.
The term ‘supercoiling’ literally means “the coiling of a coil”: the cord of a landline telephone, for example, is a coiled wire; when the coiled wire is over- or under-coiled compared to what we could call a “stable” number of turns, it twists over itself due to the strain caused, and we can describe that as a “supercoil”.


(Credit: groundguitar.com)
Likewise, DNA supercoiling refers to the over- or underwinding of a DNA strand caused by the strain on it, and to the way the DNA helix turns over itself in order to release the tension. In a “relaxed” double helix segment of B-DNA (as a reminder, there are three major forms of double-strand DNA in which single strands are connected by interactions between complementary base pairs: A-form, B-form,and Z-form DNA; B-DNA is the double helix described by Watson and Crick, and is the most common in cells), the two strands twist around the helical axis once every 10.4-10.5 base pairs. As twists are added or removed, changes to the strain of the DNA double helix are created and it causes a supercoil.3
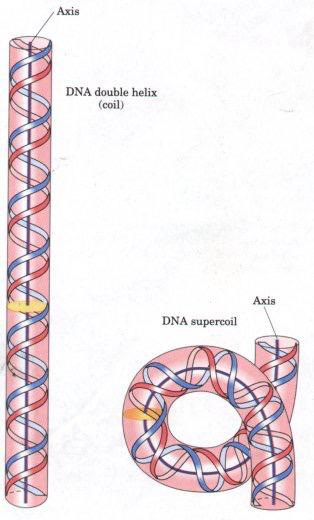
But these changes do not happen at random; on the contrary, the supercoiling of the DNA double helix is modulated according to the cell cycle phase and transcriptional activity,4 and every change is strictly controlled as each one of them regulates access to the genetic code, and may affect metabolism and gene expression. Certain enzymes known as topoisomerases are involved in the regulation of DNA supercoiling: they are required to avoid superhelical tension and knots in order to maintain DNA compacted, and they are able to change the degree of supercoiling without altering the structure or sequence of nucleotides.5
During transcription and replication, the DNA needs to be unwound in order for the transcription/replication machinery to gain access to the DNA so it can be copied or replicated. As we can expect with from what we know now, unwinding will lead to a change in the strain, and this will subsequently cause a change in the way DNA is supercoiled. Topoisomerases play a role here. For example, type I topoisomerases cause single-strand breaks and re-ligation when the DNA helix must be unwound, and type II topoisomerases cause double-strand breaks6 that are subsequently resealed after changing the twisting status of the double helix.
It is not difficult to imagine that, since topoisomerases regulate processes related to DNA replication, they are interesting as drug targets, and in fact their study has given raise to many antibiotics and anticancer drugs.2As an example, the mechanism of action of the broad-spectrum fluoroquinolone antibiotics consists in efficiently disrupting the function of bacterial type II topoisomerases by preventing them from creating breaks in chromosomal DNA when needed, thus hijacking the transcription and replication of bacterial DNA.
Another interesting example is the interaction of some drugs with topoisomerase type IV, that generates double-stranded breaks in chromosomes. Despite being essential for cell survival, this enzyme has the potential to fragment the genome if it works more than required,7 and this potentially lethal characteristic is exactly what quinolones take advantage of: their mechanism of action consists on killing bacteria by uncontrollably increasing the concentration of enzyme−DNA complexes which cause the bacterial DNA to break. These type of antibiotics are also known as “topoisomerase poisons” because they convert topoisomerases into cellular toxins.8
An increasing number of anti-cancer drugs work by interfering with the functions of topoisomerases type I and II in malignant cells. These chemotherapies inhibit the enzymes by blocking the reaction that reseals the breaks in the DNA, creating a permanent breakage in it that leads to cell death (apoptosis). Most of these drugs are selective against either topoisomerase I or II, but some can target both enzymes.
Topoisomerase I inhibitors induce single-strand breaks into DNA, and can work by a variety of mechanisms: they can cause chromosomal aberrations, and some drugs, such as camptothecins, stabilize the complex formed by topoisomerase I and DNA by inhibiting its dissociation; once they are stuck together this leads to DNA damage.
Topoisomerase II inhibitors, like topoisomerase type I inhibitors, can cause chromosomal aberrations, and can act by either stabilising topoisomerase II-DNA complexes that would be otherwise easily cleaved, or by interfering with the catalytic activity of the enzyme, both resulting in double-strand breaks in the DNA. Anthracyclines are topoisimerase type II inhibitors that are amongst the most widely used anti-cancer agents. These drugs induce double -trand breaks in DNA, and cause arrest in the cell cycle by disrupting the interaction between topoisomerase II and regulators of the cell growth. They act by stabilizing a reaction intermediate in which DNA strands are cut and linked to topoisomerase II, eventually impeding DNA resealing and preventing the cancer cells from continuing their normal life cycle.9
There are also dual inhibitors that target both topoisomerase I and II at the same time, which increases the potency of the anti-cancer effect. These drugs work by a variety of means: by recognising structural motifs present on both enzymes, by linking separate topoisomerase inhibitors together into a hybrid drug, or by using inhibitors that bind to DNA and intercalate it.
An interesting feature of inhibitors of both topoisomerase types I and II, is that they exploit the fact that DNA damage can be repaired more efficiently in normal cells than in cancer cells since the latter are deficient for DNA repair. While a deficient DNA damage response is a typical characteristic of cancer cells10 (and what ultimately causes cancer due to it allowing abnormal cells to replicate and eventually causing malignant tumours), the lack of DNA repair mechanisms can be exploited in these cases, in order to damage those same malignant cells in a more efficient manner.11
References:
1Annunziato, A. “DNA Packaging: Nucleosomes and Chromatin”. Nature Education 2008, 26.
2Pommier, Y., et al. “DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs”. Chemistry & Biology 2010, 421.
3D.M.J. Lilley, in Encyclopaedia of Genetics, 2001.
4Binaschi M., et al. “Anthracyclines: selected new developments”. Current Medicinal Chemistry2001, 1, 113.
5Champoux, J. J. “DNA topoisomerases: structure, function, and mechanism”. Annual Review of Biochemistry 2001,70, 369. doi:10.1146/annurev.biochem.70.1.369.
6Bar, A. et al. “Denaturation of circular DNA: Supercoil mechanism”. Physical Review E. 2011, 84 (4), 041935-1. doi:10.1103/physreve.84.041935.
7 Aldred, K. J. et al. “Mechanism of Quinolone Action and Resistance”. Biochemistry 2014, 53, 1565. doi: dx.doi.org/10.1021/bi5000564 |
8Kreuzer, K. N., and Cozzarelli, N. R. “Escherichia coli mutants thermosensitive for deoxyribonucleic acid gyrase subunit A: Effects on deoxyribonucleic acid replication, transcription, and bacteriophage growth”. Journal of Bacteriology 1979, 140, 424.
9Minotti, G., et al. “Anthracyclines: Molecular Advances and Pharmacologic Developments in Antitumor Activity and Cardiotoxicity”. Pharmacology Reviews 2004, 56, 185.
10Hanahan, D. and Weinberg, R. A. “Hallmarks of cancer: the next generation”. Cell 2011, 144, 646.
11Van Gent, D. and Kanaar, R. “Exploiting DNA repair defects for novel cancer therapies”. Molecular Biology of the Cell 2016, 27 (14), 2145.
3 comments
[…] diseinuan aprobetxa daitezke konplexutasun eta paketarzen horiek. Isabel Pérez Castrok azaltzen du Exploiting DNA packaging […]
[…] El ADN humano tiene un empaquetamineto tan compacto y complejo que toda esa información cabe un núcleo celular que apenas tiene 6 micras de diámetro. Esa complejidad y compactificación pueden aprovecharse en el diseño de fármacos. Isabel Pérez castro […]
[…] El ADN humano tiene un empaquetamineto tan compacto y complejo que toda esa información cabe un núcleo celular que apenas tiene 6 micras de diámetro. Esa complejidad y compactificación pueden aprovecharse en el diseño de fármacos. Isabel Pérez castro […]