The ultrafast dynamics of adsorbates deciphered
The ultrafast dynamics of adsorbates deciphered
One of the ultimate goals in surface science is to comprehend the fundamental processes that make that surface reactions need less than a picosecond (10-12 s) to occur. This means understanding what is happening at the scale of femtoseconds, that is, measuring things that take some tens or hundreds of 10-15 seconds to occur.
To acquire such a time-resolved insight, numerous experiments have studied the ultrafast elementary motions of adsorbates on metal surfaces, such as vibrational motion, molecular desorption, diffusion, or dissociation, by means of time-dependent techniques. In all these experiments, the intense femtosecond laser pulses initiates the energy exchange mechanisms between the laser-excited surface electrons and the vibrational modes of both the adsorbates and the surface lattice.
In time-resolved infrared (IR) spectroscopy experiments, the initial adsorbate dynamics started by the pulse is directly probed by tracking the frequency shift and linewidth changes of the so-called IR-active internal stretch (IS) mode. In all adsorbate-surface systems investigated thus far, the IS frequency mode exhibits an initial redshift followed by a rapid blueshift. Why this happens, nobody knows for sure.
Even though there have been some speculation about the origin of this behaviour, the fact is that the lack of a general quantitative theory effectively prevents us from using time-resolved vibrational spectroscopy to its full potential or, from the general surface science perspective, from extracting the critical information about what is happening in less than a picosecond.
In order to solve this problem, a team that includes researchers from CFM, DIPC and UPV/EHU, has created 1 a general first-principles theoretical framework that makes possible to calculate directly the vibrational spectra changes and identify the specific mechanisms behind them.
The new theory relies on a recently developed approach based on many-body and density functional perturbation theories that the team extends now to to treat nonequilibrium conditions. The first-principles formalism accounts for electron-phonon coupling processes up to second order, including electron-hole pair excitations (the first-order non-adiabatic coupling) as well as vibrational intermode coupling due to the indirect interaction with hot electrons.
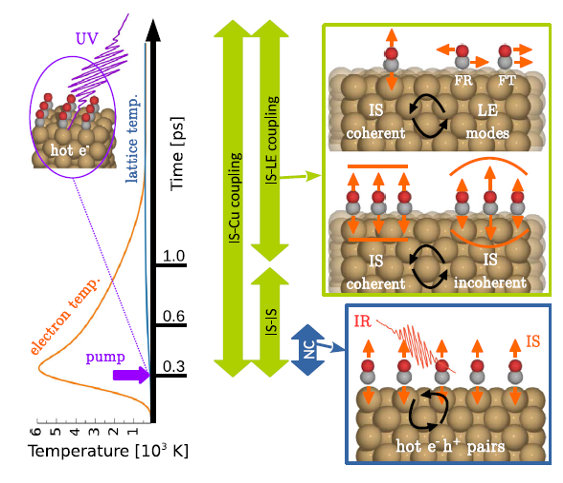
As a proof of concept, the team applies this theoretical framework to investigate the early stage dynamics of the benchmark system for electron-induced dynamics at surfaces, non-thermal CO adsorbates (i.e., a molecular overlayer) on Cu(100). This system has recently been monitored with time-resolved sum-frequency generation spectroscopy with unprecedented subpicosecond resolution. It is the new quantitative and predictive theoretical method which finally unveils the microscopic processes behind the reported non-thermal frequency shifts and the accompanying linewidth changes. It also establishes the specific sequence and strength of the non-adiabatic and intermode coupling mechanisms involved.
The researchers find that, contrary to common understanding, the CO internal stretch mode undergoes different mode coupling mechanisms on the subpicosecond and the picosecond timescales. It turns out that the C-O interaction, which is in the end responsible for the IS frequency, is more sensitive to the transient excitations created in the electronic system, while it is the coupling to the excited phonon modes that contributes more to the IS linewidth.
This new theory is destined to be the theoretical counterpart in future vibrational spectroscopy investigations. The time-resolved nanoscopic insights that this theory can provide would be, not only fundamental to the development of vibrational spectroscopy at surfaces, but the reference to advance in our goal of controlling surface reactions at the molecular level.
Author: César Tomé López is a science writer and the editor of Mapping Ignorance.
References
- D. Novko, J. C. Tremblay, M. Alducin, and J. I. Juaristi (2019) Ultrafast Transient Dynamics of Adsorbates on Surfaces Deciphered: The Case of CO on Cu(100) Phys. Rev. Lett. doi: 10.1103/PhysRevLett.122.016806 ↩