The relationship between molecule positioning and friction at the atomic level
The relationship between molecule positioning and friction at the atomic level
Friction, an everyday phenomenon, has perplexed scientists for centuries. Though extensively researched, our understanding remains fragmented, primarily due to the multifaceted interactions that span across varying scales. Achieving an accurate grasp of the precise contact conditions between objects has been a longstanding challenge, a feat recently made possible through advancements in scanning probe microscopy.
Yet, even with these technological breakthroughs, the intricacies of dynamic friction – the force needed to keep sliding – have remained elusive. While scientists could measure static friction by moving a single molecule on a surface, both the measurement and theoretical understanding of dynamic friction have yet to be fully unveiled.
Friction is an elusive phenomenon because it arises from the interplay of several physical mechanisms typically spanning very different length scales. Seeking to isolate these, research on friction has reached the atomic level in the past decades owing to the progress of experimental techniques represented by scanning tunneling spectroscopy (STM) and atomic force microscopy (AFM), and by computer simulations. This way, the empirical laws observed at the macroscopic scale have been linked to processes at the nanoscale.
The most elementary approach to study friction would be a single atom moving over a surface with STM and AFM. These techniques are primarily used to visualize single atoms and molecules on surfaces, and even allow to manipulate them by the interaction force from the tip, enabling the fabrication of fascinating structures such as quantum corrals, computing devices and molecular graphene. Controlled manipulation of atoms with STM/AFM at liquid helium temperatures provides a powerful means to investigate friction and lubricity at the fundamental level in a highly reproducible way, as the system consists of only three bodies involving two atomic-scale contacts where thermal diffusion effects can be minimized. In addition, the geometry of the surface and tip apex as well as the adsorption site of the atom can all be determined before and after the manipulation.
A remarkable result obtained this way was the measurement of the lateral force needed to initiate sliding of single atoms, i.e. the determination of the static friction force. However, insight into dynamic friction (force needed to keep sliding) at the atomic scale requires a complete picture of the dynamics, involving intermediate states and energy dissipation, which is missing so far.
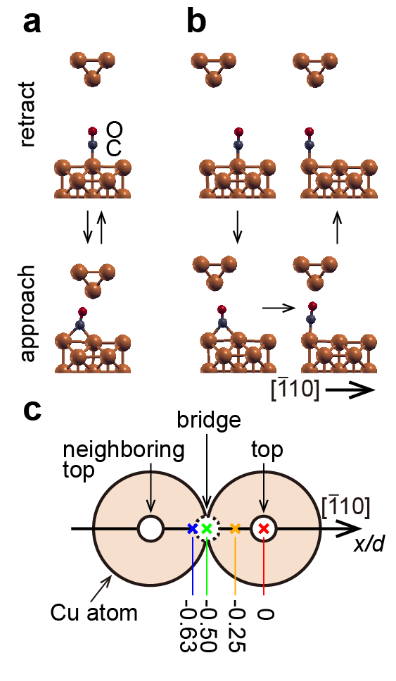
Now, a team of researchers reports 12 a groundbreaking study that dives deep into this challenge. The researchers meticulously examined the manipulation of a carbon monoxide (CO) molecule on a single-crystal copper (110) surface using an AFM. Backed by ab initio calculations, their findings shed light on how the CO molecule positions relative to the microscope tip and surface, and the relationship between molecule positioning, energy dissipation, and both static and dynamic friction.
The key is the discovery of an unexpected intermediate state that reshapes our foundational understanding of the process. Imagine a billiard table covered with balls that are closely packed together. This represents a surface which is not completely flat at the atomic scale. Normally, when we deposit a molecule on the surface, it sits comfortably atop a single ball. But when pushed across this ‘table’, instead of hopping directly from one ball to the next, the molecule sometimes takes a surprising detour, momentarily positioning itself between two balls.
Thus, when the AFM tip is located far from the substrate, CO adsorbs on a top site in an upright configuration with its C atom bound to the Cu atom. By approaching the metallic tip over the top site to a close enough distance, CO moves along the [110] direction to a bridge site. Bringing a vertically oscillating tip close to the surface directly over the CO molecule leads to correlated lateral jumps of the CO molecule between top and bridge sites.
When the oscillating tip approaches on a laterally shifted location closer to one of the neighboring top sites, the CO molecule is also manipulated from top to bridge site. However, in this case the CO molecule irreversibly ends up in the neighboring top site that is closer to the metal tip apex when the tip retracts.
This intermediate state, inaccessible for the far-tip position, is enabled in the reaction pathway for the close-tip position, which is crucial to understanding the manipulation process, including dynamic friction. In order to understand the energy dissipation that occurs during these manipulation processes, the scientists calculated the potential energy between model tip structures and a CO on Cu(110) as a function of lateral and vertical tip position.
This unexpected ‘stopover’ challenges our previous understanding of how the molecule moves. The results show how friction forces can be controlled and optimized, facilitating new fundamental insights for tribology.
Author: César Tomé López is a science writer and the editor of Mapping Ignorance
Disclaimer: Parts of this article may have been copied verbatim or almost verbatim from the referenced research paper/s.
References
- Norio Okabayashi, Thomas Frederiksen, Alexander Liebig, and Franz J. Giessibl. (2023) Dynamic Friction Unraveled by Observing an Unexpected Intermediate State in Controlled Molecular Manipulation Phys. Rev. Lett. doi: 10.1103/PhysRevLett.131.148001 ↩
- Norio Okabayashi, Thomas Frederiksen, Alexander Liebig, and Franz J. Giessibl (2023) Energy dissipation of a carbon monoxide molecule manipulated using a metallic tip on copper surfaces Phys. Rev. B doi: 10.1103/PhysRevB.108.165401 ↩