The 3D structure of the genome
The 3D structure of the genome
The genome is the text of life. Its ink is the deoxyribonucleic acid (DNA), a heteropolimer of four nucleotides (guanine, adenine, thymine, and cytosine). It encodes the instructions to make work all known living organisms (and viruses). It was isolated at first in 1869 for Friedrich Miescher and at the early 50s of the 20th century was confirmed as the material containing genetic inheritance. Just afterwards, James D. Watson and Francis Crick suggested the first double-helix model of DNA structure (Fig. 1A). It is known for long that the double-stranded DNA self-organizes hierarchically by protein interaction, giving the chromatin the higher order structural features, classified as primary, secondary and tertiary (Fig. 1B) (Do not confuse with primary, secondary and tertiary structure of DNA double-helix, where chromatin level is considered the quaternary structure). These chromatin features are highly dynamic, with non-random distribution, varying during the cell cycle and between different cell types. Plenty of evidences show that these higher order structural features are also paramount in gene expression and genetic recombination.
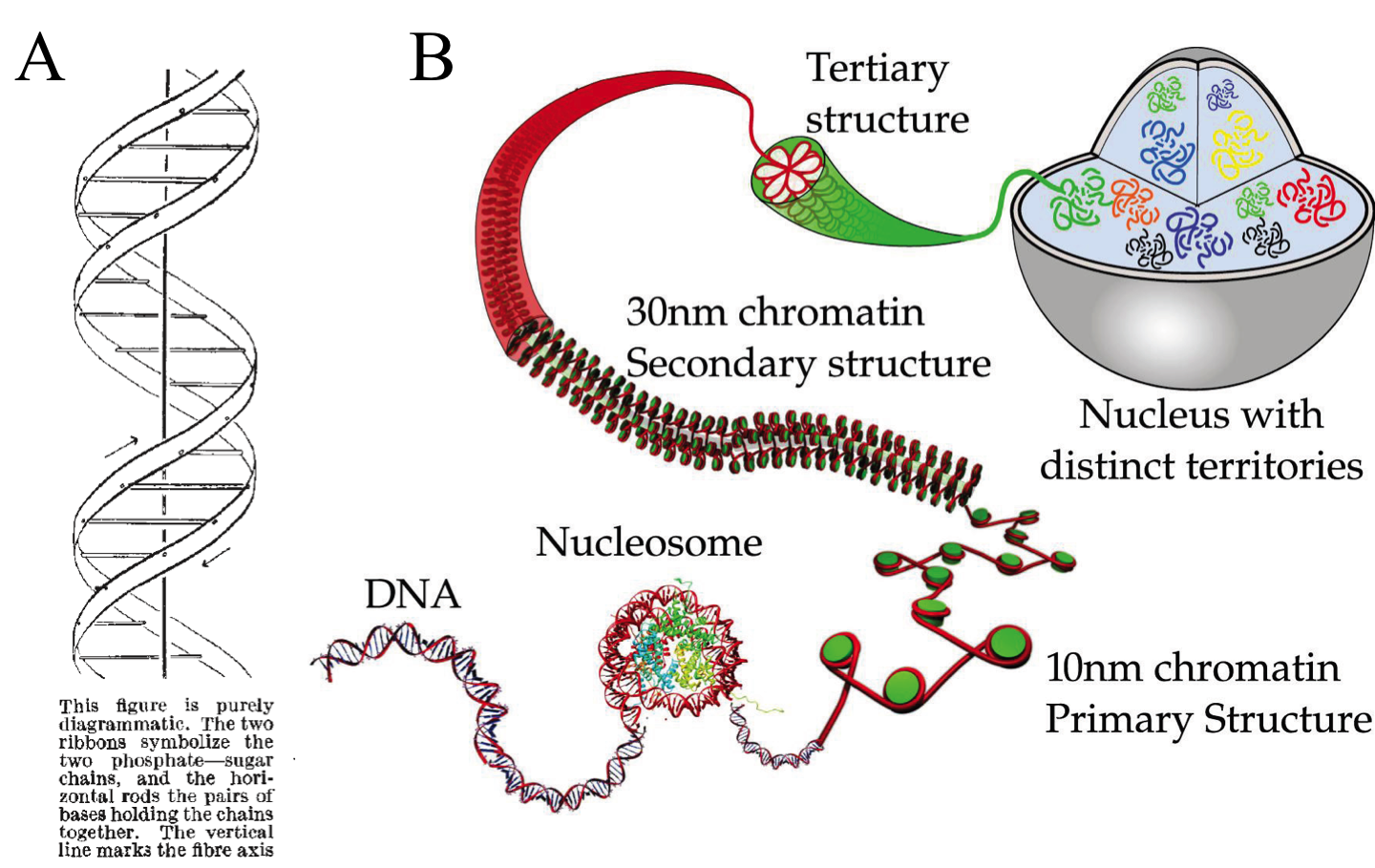
Note that chromatin compaction at tertiary level can be divided into two groups depending on the phase of the cell cycle. During interphase, while the cell perform its normal activities, the tertiary structure is arranged at gene locus level. During metaphase, where cell division takes place, the tertiary structure is organized forming chromosomes. They vary from 50 to 100,000-fold compaction respectively. Mapping the chromosomes tertiary structure during interphase is key to understand gene regulation underlying processes (transcription, translation, DNA repair and recombination), which seems to be spatially compartmentalized and not ubiquitous.
For years, some techniques like electron and light microscopy, fluorescence in situ hybridization (FISH) and chromosome regions specific labelling were the spearhead finding and defining chromatin conformation and structure (Fig. 2A). However, they had no power to define the complete three-dimensional maps. They were able to show us how were a house or a neighborhood but unable to locate them in the country. Job Dekker and coworkers developed in 2002 a high-throughput methodology called Chromosome Conformation Capture (3C). They used Saccharomyces cerevisiae isolated nuclei and subject them to formaldehyde fixation. The formaldehyde is a chemical cross-linker affecting physically close segments of DNA complexed with proteins. Then, the analysis of relative frequencies with which different sites (detected by PCR) have become cross-linked provides information about general chromatin organization, physical properties and conformations of chromosomes (Fig. 2B).

Such an idea has been exploited in the last decade through successive stepwise implementations and refinements to finally face genome-wide analysis. The early versions, even been high-throwput, required to choose a set of target loci or single chromosomes, and hence a still reduced genome region. However, none of them, including the last ones, are able to directly measure the 3D-distances. But it does not preclude that, by combining interaction data sets with known distances between sub-nuclear landmarks and computer simulations, can be defined robust 3D models of the entire genome. Using Chromosome Conformation capture-on-Chip (4C) coupled to a massive parallel sequencing, Duan and colleges have constructed a three-dimensional model of the yeast genome 1 (Fig. 3). Furthermore, Lieberman-Aiden and co-workers, headed by Dekker, have recently summarized all the implementations in one to bring what seems to be the more powerful improvement of this methodology up to date, called Hi-C. It built spatial proximity maps of the human genome. They show that, unlike the more commonly used globular equilibrium model with highly entangled structure where neighbouring loci do not need to be nearby in 3D; the local packing of human chromatin is consistent with the behaviour of a fractal globule, where neighbouring loci tend to be nearby in 3D. In addition, it confirms the existence of chromosome territories and the spatial proximity of gene-rich chromosomes and identifies two genome-wide compartments separating open (accessible, actively transcribed) and closed (densely packed, less transcribed) chromatin (Fig.3B).
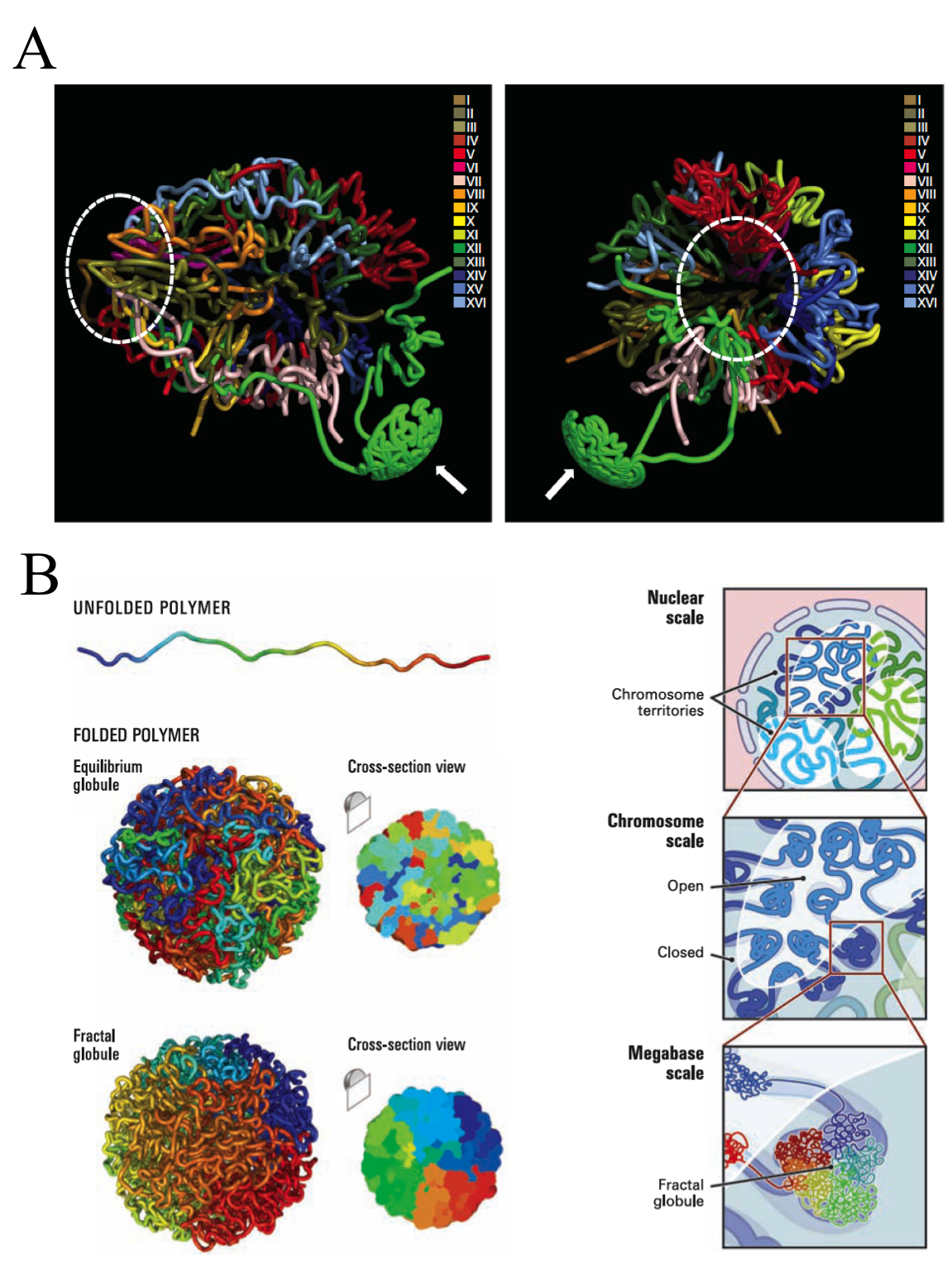
These findings are good examples about how a simple idea implemented with high-throwput technologies allows us to cover very wide ranges of scale, answering questions otherwise intractables. It is known long ago the existence of regulons in yeast. They are entities formed by genes or operators distributed in the genome that are expresed under the same control events and regulatory proteins. So early, this common regulation of genes, which are located far away of each other in the chromosome sequence, suggested that some complex chromatin structure should exist underlying the gene activity. In fact, microscopy and chromosome region specific labelling provided some solid evidences at this respect. Now these new high-throwput technologies confirm some features and expand our understanding revealing subtle but important details about the chromatin structure. And aditionaly, they contribute to figure up hidden mechanisms involved in some human illneses. In the case of some kinds of cancer, an extended feature is that after double-strand DNA breaks, the incorrect reparation of two initially non-sequential fragments of DNA become sequential generating translocation events. The hypothesis suggests that translocation happens between DNA strans colocalizing in nuclei of normal cells before the pathologycal rearrangement. As proof of art, Hi-C provide further evidence about that hypotesis and allow genome-wide analysis of structural rearrangements of a cancer cell 2. The later knowledge could open new ways to completely understand the illness and, therefore, new ways to face it.
References
- Duan Z., et al. 2010. A three-dimensional model of the yeast genome. Nature 465 20 May. doi:10.1038/nature08973 ↩
- Engreitz JM, Agarwala V, Mirny LA (2012) Three-Dimensional Genome Architecture Influences Partner Selection for Chromosomal Translocations in Human Disease. PLoS ONE 7(9): e44196. doi:10.1371/journal.pone.0044196 ↩
4 comments
[…] Artículo original publicado por Daniel Moreno Andrés en Mapping Ignorance […]
[…] of the replication-timing program. In fact, using the Hi-C (find more info about this technique here), It was observed that those genome regions that typically replicate simultaneously tend to be […]
this is very useful n knwlagable … it provides such an important fetures of bio. it increases information.
[…] Figura 1 – Rappresentazione grafica dell’organizzazione del DNA nella cellula. [Fonte: The 3D structure of the genome – Mapping Ignorance] […]