Mutations in the non-coding genome contribute to autism
Mutations in the non-coding genome contribute to autism
A significant portion of an organism’s genome is non-coding; that is, it does not carry information to make proteins. Part of the non-coding DNA is transcribed into functional non-coding RNA molecules (e.g., transfer RNA, ribosomal RNA and regulatory RNA). Other functions of non-coding DNA include transcriptional and translational regulation of protein coding sequences, nuclear scaffold binding regions, DNA replication origins, centromeres and telomeres.
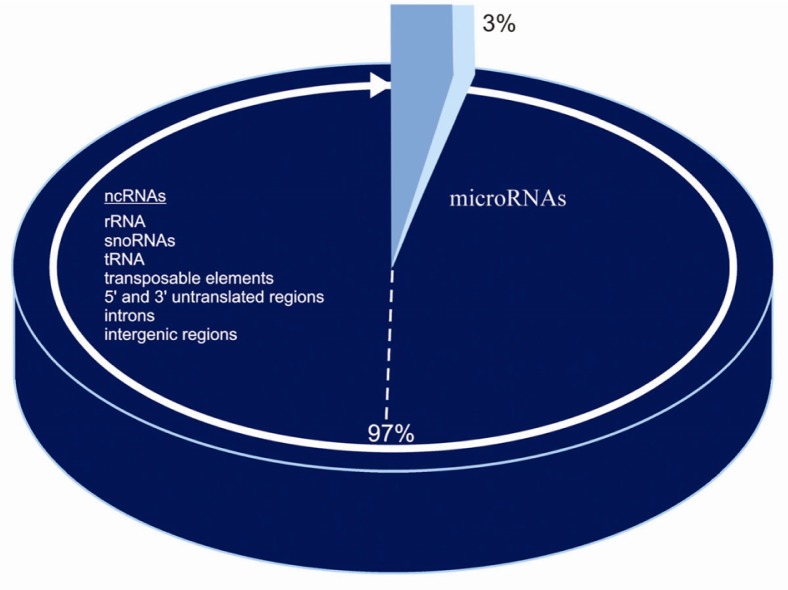
Coding DNA is approximately 3% in the human species, while the non-coding genome is the remaining 97%. It is a mistake to call non-coding DNA junk DNA because it is involved in epigenetic activity and complex networks of genetic interactions; in simple words, when, where and how much genes are expressed 1.
Unlike coding segments (exons), the boundaries of non-coding regions are not well-defined and therefore different types of marks such as evolutionary conservation, sequence restriction and epigenetic marks are useful guides for their identification and analysis.
A recent study whose first author is Evin Padhi 2 has shown that spontaneous mutations in non-coding regions of the genome are associated with autism. Thus, the genetics of autism is not limited to coding genes, but non-coding DNA also influences and may explain the autism of some affected individuals.
Statistical studies had suggested that non-coding segments had variants that contributed to autism, and this study is the first to identify three individuals with mutations in the non-coding genome. It seems clear that in the near future there will be a fairly extensive list of de novo variants in non-coding sequences implicated in autism, just as we already have in coding DNA.
The researchers evaluated whole genome sequencing data on 2671 families with autism (discovery cohort of 516 families, replication cohort of 2155 families). They focused the study on 544 non-coding segments of the genome known to be active in the brain of mouse embryos. These segments, termed enhancers, increase transcription of coding genes .
They then searched the whole genome sequences of the 2,671 families with an autistic child present in the Simons Simplex Collection database to see which of these enhancers contained de novo variants. That is, they first identified enhancers that act in the brain and then looked at the non-protein-coding but enhancer-encoding DNA to see if there were differences between families with autism and families without autism.
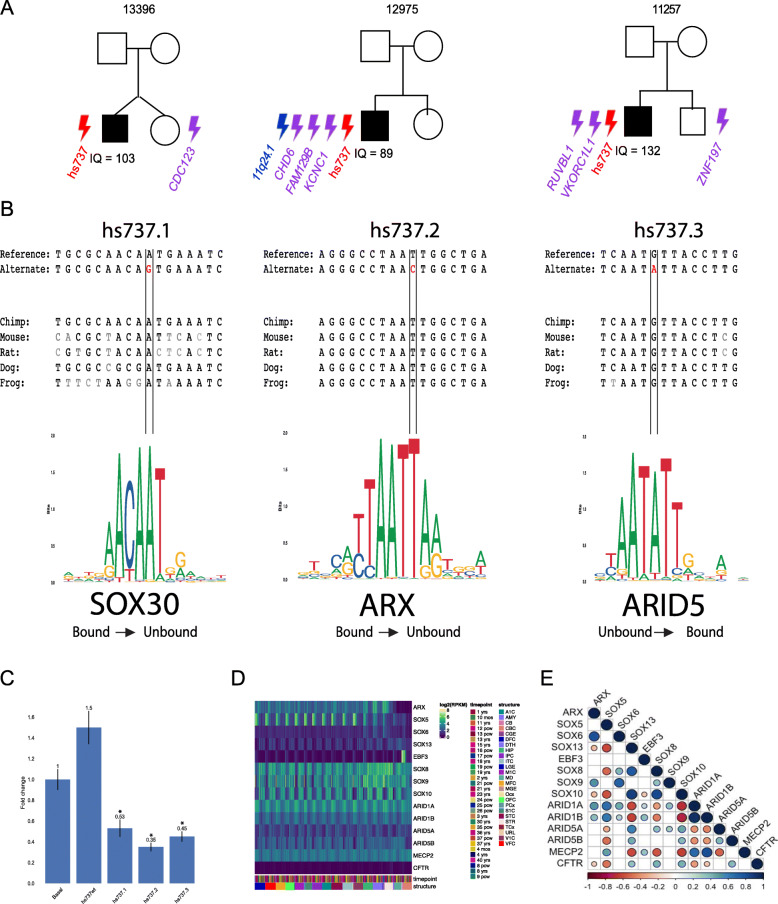
One particular enhancer, known as hs737, showed significantly more de novo variations in both the discovery cohort of 516 families and the replication cohort of 2,155 families. Notably, these de novo mutations in hs737 appeared in three individuals with autism with no other known autism-related variants; that is, individuals with autism who did not have autism-related coding genes may have autism-related non-coding genes.
Individuals with de novo variations in hs737 had shared phenotypes, commonality in being male, intact cognitive function, and hypotonia or motor delay. This contrasts with de novo variations in coding genes, which are more abundant in females with intellectual disability.
Using a neuronal cell line, the researchers saw that in all three cases, the de novo mutations caused the enhancer to reduce, not increase, gene expression. Using epigenomic analysis, the researchers were able to see that hs737 is brain-specific and acts on the EBF3 transcription factor gene in the human fetal brain.
To the researchers’ surprise, EBF3 is a well-studied gene. Mutations in EBF3 cause a condition called hypotonia, ataxia and developmental delay syndrome (HADDS), characterized by low muscle tone and motor problems. EBF3 has de novo variations in neurodevelopmental disorders and is widely expressed in the body. The outcome is not similar in cases where the EBF3-related de novo variations (DNVs) are in either coding or non-coding areas. Individuals with coding DNVs have greater phenotypic severity (hypotonia, ataxia, and delayed development syndrome) compared to individuals with non-coding DNVs who have autism and hypotonia.
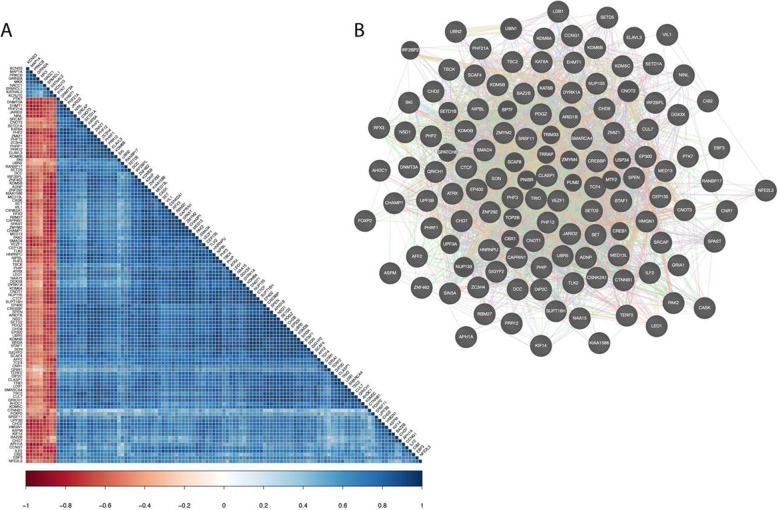
The researchers analyzed EBF3 binding to promoters and identified 3100 genes (16% of all genes in the genome) to which EBF3 binds. They then focused on genes encoding VDNs in neurodevelopmental disorders and to which EBF3 bound. Of the 253 genes implicated in neurodevelopmental disorders described by Coe et al. Coe BP, Stessman HAF, Sulovari A, Geisheker MR, Bakken TE, Lake AM, Dougherty JD, Lein ES, Hormozdiari F, Bernier RA, Eichler EE (2019) Neurodevelopmental disease genes implicated by de novo mutation and copy number variation morbidity. Nat Genet 51(1): 106-116. doi: 10.1038/s41588-018-0288-4.[/foornote], 26.9% of them had EBF3 binding to their promoter. These DNVs are found in highly conserved nucleotides and are predicted to affect transcription factor binding at the enhancer.
The hs737 is only active in the brain while the embryo is developing, so mutations in the enhancer could result in similar but less severe traits than mutations in the gene itself. In other words, if you affect a coding gene the effect can be total and forceful, if you affect the enhancer of that gene the effect might not be as severe. This would also explain the presence of the spectrum: mutations in a coding gene can make it functional or not, mutations in a non-coding DNA (regulator) can theoretically make a gene express 30, 70 or 150% of normal.
A key problem in the study of enhancers is determining which gene they regulate. It is especially tricky when the enhancers are far removed from the promoter they target, but there are tricks to achieve this. EBF3 is a transcription factor that is ubiquitously expressed in humans, affects many genes and, in particular, in neuronal cells. Regulatory genes form a broad network in which one regulates another. The higher the location of the variant in the network, the more severe the phenotypic consequence and the lower in the network, the less severe the phenotypic consequence (e.g., mutation in the hs737 enhancer). EBF3 regulates other genes involved in down-regulation in neurodevelopmental disorders.
The researchers tested many sequences, but only one enhancer achieved a difference that falls within the level of significance established in this study. However, it seems clear that other regulators will be found in which a link to autism will also be seen. Finally, Padhi’s group is working on developing a strain of mice lacking the hs737 enhancer, in order to better understand how mutations in that non-coding DNA sequence affect behavior (1).
References
- Schenkman L (2021) Mutations in the noncoding genome contribute to autism. Spectrum Aug. 2. https://www.spectrumnews.org/news/mutations-in-the-noncoding-genome-contribute-to-autism/ https://doi.org/10.53053/VCFT1127 ↩
- Padhi EM, Hayeck TJ, Cheng Z, Chatterjee S, Mannion BJ, Byrska-Bishop M, Willems M, Pinson L, Redon S, Benech C, Uguen K, Audebert-Bellanger S, Le Marechal C, Férec C, Efthymiou S, Rahman F, Maqbool S, Maroofian R, Houlden H, Musunuri R, Narzisi G, Abhyankar A, Hunter RD, Akiyama J, Fries LE, Ng JK, Mehinovic E, Stong N, Allen AS, Dickel DE, Bernier RA, Gorkin DU, Pennacchio LA, Zody MC, Turner TN (2021) Coding and noncoding variants in EBF3 are involved in HADDS and simplex autism. Hum Genomics 15(1): 44. doi: 10.1186/s40246-021-00342-3. ↩