Computation can push optical microscopy towards unsuspected limits
Computation can push optical microscopy towards unsuspected limits

Man does not live by hardware alone. Indeed, great material and conceptual improvements in the machinery of optical microscopes have occurred in recent decades. The examples are numerous (some example here; https://mappingignorance.org/2013/12/23/bessel-beam-plane-illumination-microscopy-another-smart-solution-for-an-old-challenge/). However, what is being achieved only with software and computing power seems a matter of magic. It is not only that programs and computers controlling microscopes are faster, more reliable and able to store and handle amounts of data orders of magnitude greater than before. Neither that improvements in hardware come together with improvements and new features in software. It is rather that the mere implementation of certain advanced computer programmings, like machine-learning algorithms and deep learning in artificial neural networks, are taking optical microscopy out of its physical limits. Here, I point out two of those recent software-based advances that can transform a decent but modest optical microscope, without additional changes in the instrument, on something that can accomplish outputs well beyond its physical capabilities.
In optical microscopy, a more or less complicated optical system control visible light to generate a magnified image of a tiny sample. This is true just when the sample or its parts have enough contrast to differentiate them from the background. However, most of the cells and tissues from living matter are quite transparent, showing just a few translucent or dense structures when placed under the transmitted light of a microscope (like bright-field illumination, the simplest of the illuminations). To circumvent the problem three basic but differentiated instrumental approaches have been developed over the last 150 years, with an infinity of modalities within each one: histological staining, phase contrast microscopy and fluorescence microscopy.
Fluorescence microscopy using widefield (low resolution) or confocal (high resolution; diffraction-limited) microscopes has become one of the most appreciated workhorses on life sciences. Leaving other kinds of staining technics and different bright-field and phase-contrast modalities for more preliminary or unusual experimental settings. For compelling reasons as higher resolution and specificity when imaging microscopic structures, the balance shifted towards fluorescence technics in detriment of other ways of increasing the contrast in the sample. Manifold approaches on fluorescence microscopy allow seeing, tracking and analyzing single subcellular structures in fixed and living cell samples with astonishing clarity and precision. For this, building blocks of those target structures have to be labelled with fluorescent molecules. Today there are dozens of ways to do this. Once this is achieved, illuminating the sample under the microscope with the appropriate light wavelength to stimulate the fluorophore will rebel the target structure as a fluorescent shape, shining over a black background. Several different fluorophores able to emit light of different wavelength can be combined (up to four usually) to reveal several different subcellular structures at a time. If required, this can be done on top of bright-field or phase-contrast imaging, which use to be the default light path in an optical microscope.
However, the nature of the light wavelengths needed to make shine those fluorophores, the nature of the fluorophores and the labelling processes bring several drawbacks. On one side, the fluorescence types of equipment and labelling materials are expensive in money and time. On the other side, performing fluorescence microscopy successfully requires a compromise between fluorophore brightness, light exposure, spatial resolution and imaging speed to avoid damaging the sample. For example, long-term imaging of living samples with a high temporal and spatial resolution just to result in fluorophore bleaching and phototoxicity, which is very harmful to cells. Rendering very soon useless samples and very low-quality images. These undesirable effects are caused by the particular illumination needs: higher light exposure (more power and/or during more acquisition time to increase the signal to noise ratio; SNR) and higher frame rates during long periods.
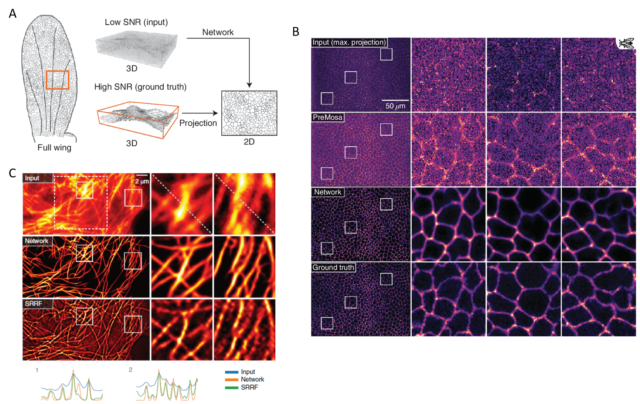
Therefore, frequently one must renounce to some dimension in the image acquisition to reach great quality in the others. Alternatively, a long process of optimization will be needed to find an intermediate point in all of them that allows collecting the necessary data for a specific purpose. Yet that is not always possible, and is here were computational approaches will seemingly provide unsuspected solutions. For example, the new content-aware image restoration (CARE) method based on convolutional neural networks allows obtaining high-resolution images from several completely different specimens using diverse approaches of fluorescence microscopy but using 10 to 60 times less light 1. This means that this computational approach can obtain high SNR images while using much less light power and/or shorter acquisition times, which avoid phototoxicity and bleaching. Besides, the time saved in the acquisition of the images facilitates imaging higher frame rates during longer times without damaging the sample.
Good magic always has a good trick. In summary, the trick with CARE is that the images are captured with settings (low power/short acquisition time) that produce low SNR, which yield images of very low quality. Then, the neural network can clean and correct those deficient images to convert them into high-resolution ones. To obtain those high-quality restored images, the neural network is trained beforehand with matched pares of low and high SNR fluorescence images. With this, the neural network “studies” how a high-quality image looks like if it would be acquired with low light power and/or short exposure time. Then, with what the neural network has learned, it can calculate and restore useful high-quality images from other images captured only at low quality, which would be useless to extract any biological conclusion (Figure 1 a, b). Such an approach, with the due modifications in the neural network, in the models and programming, can cope with many different imaging problems. Getting even to resolve sub-diffraction structures (separated less than 200nm) using only low-resolution fluorescence images from widefield microscopy as input (Figure 1 c). However, since the restorations are non-linear, the intensity quantitation approaches (e.g.: for estimating protein amounts) on the output image are not valid. Additionally, incompatibilities on the restoration may appear when the sample contains biological structures that are not present in the training set.
Another recently developed tool based on neural networks to overcome some of the drawbacks of the fluorescence microscopy can recognize and extract subcellular structures on bright-field images 2. As pointed out above, the light sources needed for illuminating properly the samples in fluorescence microscopy are very energetic and, potentially, quite dangerous for the integrity of the sample. However, transmitted light illumination (bright-field, phase contrast…), which illuminates the sample with low power light in the visible region of the light spectrum, is harmless to the specimen. In counterpart, subcellular structures display no or extremely low contrast, which makes difficult to charge slight changes on the visible subcellular changes even to trained biologists. In this case, the neural network is trained with paired bright-field and fluorescence images of cell layers with fluorescently labelled subcellular structures and organelles. Here the network learns how the low contrast shapes in the bright-field image correlate with the accurate ground-truth stablished by fluorescently labelled structures. Then, the network generates models that can predict the distribution, volume and shape of different subcellular structures imaged only with transmitted light. The accuracy on the prediction depends critically on how extensive has been the training of the network, and if a robust correlation can be stabilized within a given structure in transmitted light and the fluorescent image. For those structures with very low contrast when using transmitted light, the performance decrease considerably and, for the same reason than above, intensity quantitation on the output images is not reliable. However, the prediction of distribution, shape and volume has very high accuracy for most of the main subcellular compartments and organelles.
Image analysis is becoming one of the fundamental skills in life sciences. Endless algorithms and computer programs infiltrated in life sciences not long ago to extract unbiased data from images were humans fail in impartiality, in recognising patterns and where doing it manually would take forever. Those early approaches have rapidly evolved and incorporated the new artificial intelligence technologies opening now previously unseen possibilities of refinement and power. What presented here is a sample of some of the last flowers in the garden dedicated to increasing spatial and temporal resolution in optical microscopy. These software developments are in the beginning and have still very limited range of application. However, these two computer programmings approached here can seemingly solve few problems (with some restrictions) that traditionally have been solved with very expensive, not easy-to-handle machinery. On the contrary, many of these programs are open software platforms easily accessible to the research community. Additionally, they can be eventually launched in a given laboratory by any researcher with little advanced computer skills, a bit of time and maybe a bit of help. This will likely turn these powerful advanced deconvolution programmings in basic research tools. Because they will be within reach of all those labs having a modest optical microscope and the willingness to investigate the living matter at a microscopic level beyond the limitations of their equipment.
References
- Weigert, M. et al. (2018) Content-aware image restoration: pushing the limits of fluorescence microscopy. Nat Methods 15, 1090-1097, doi: 10.1038/s41592-018-0216-7 . ↩
- Ounkomol, C., Seshamani, S., Maleckar, M. M., Collman, F. & Johnson, G. R. (2018) Label-free prediction of three-dimensional fluorescence images from transmitted-light microscopy. Nat Methods 15, 917-920, doi:10.1038/s41592-018-0111-2 . ↩
2 comments
[…] optiko normal baten datuekin adimen artifizialak lortu dezakeena. Daniel Morenok kontatzen du Computation can push optical microscopy towards unsuspected limits […]
[…] Lo que la inteligencia artificial puede conseguir con los datos de un microscopio óptico normalito parece de ciencia ficción. Nos cuenta Daniel Moreno en Computation can push optical microscopy towards unsuspected limits […]