The mystery of collagen and thyroid hormone: a detective story
The mystery of collagen and thyroid hormone: a detective story

Collagen is the newest trendy nutrient. If we search for “collagen” in the web browser, we will find a wide variety of food supplements, cosmetic products and web pages where collagen beneficial effects are highlighted. Although collagen is a mere protein, its name is currently associated to the wellness circuit and, for this reason, it has become more visible than other equally important proteins like thyroid hormone receptor or transforming growth factor beta.
Collagen is the most abundant protein within the extracellular matrix: cells such as fibroblasts produce it and it is subsequently exocytosed to the surrounding medium. Despite the explicit or implicit advertising messages, collagen is not an essential nutrient, this is, there is no need to obtain it through the diet for it is normally produced by our body.
This protein forms fibres and gives strength and consistence to the different body tissues. If we let ourselves be carried away by publicity, we run the risk of considering collagen as a “good” protein, but this would be a mistake. Proteins should not be classified as “good” or “bad”. Their effects ultimately depend on the place, the moment and the quantity.
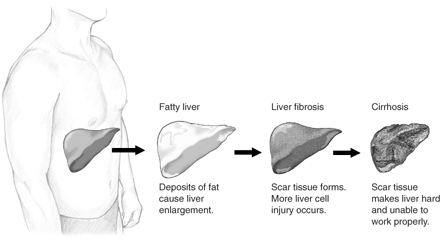
For instance, collagen is the main factor responsible for fibrosis. Fibrosis occurs as a result of an excessive deposition of extracellular matrix components and can affect different tissues. This is the base for various alterations and pathologies such as cirrhosis, cystic fibrosis, arthrofibrosis, Crohn disease, etc. This collagen accumulation does not depend on the intake of this protein on the diet.
Dr. Aranda research group have studied 1 certain molecular mechanisms that underlie the progression of fibrosis. This research may lead to antifibrotic drug design. Furthermore, these results can exemplify how to accumulate evidence in order to draw conclusions based on a vast amount of information. This is comparable to a police investigation performed at different scales (molecular, cellular and organ level), which uses a great selection of techniques.
The main characters of the investigation are the following:
-
TGFβ. This protein plays a key role in the development of fibrosis. It reaches the cell membranes and joins with its receptor. This union determines the response of the targeted cell. At the molecular level, TGFβ triggers changes in proteins located inside the cell, such as SMAD proteins.
-
SMAD. These proteins, located within the cell, are activated by a slight but still relevant chemical change: phosphorylation (addition of a phosphoryl group, a small 5-atom group). After phosphorylation, SMAD proteins enter the nucleus, where they bind certain target genes and promote collagen transcription (they are, in fact, transcription factors). The origins and the meaning of the name SMAD (small + mothers against decapentaplegic) deserve a separate note.
-
TR. The nuclear receptor on this study, which is also located inside de cell. TR recognizes thyroid hormones such as T3. When T3 and TR join together, a series of different processes is activated.
-
Fibroblasts. Ubiquitous cells in charge of the maintenance of the extracellular matrix, which is a complex network located between cells that provides structural and biochemical support to the surrounding cells.
-
Myofibroblasts. Fibroblast derived cells producing a collagen-rich extracellular matrix. Myofibroblasts participate in development of fibrosis and, unlike fibroblasts, they can express αSMA, that is, if a cell expresses αSMA it is a myofibroblast. TGFβ, one of our main characters, induces differentiation of fibroblasts into myofibroblasts.
And this is the background:
It has been shown that low levels of thyroid hormones can increase the risk of non-alcoholic fatty liver disease, which in the long term may progress to hepatic fibrosis. If fibrosis is enhanced in the absence of thyroid hormones, could these have a protective role against fibrosis?
Previous studies provided proof of interaction between TGFβ and TR pathways in liver cancer progression. Could these two factors work simultaneously to induce fibrosis?
Once the main characters, the background and the questions are fully established, it is now the time to start with the investigation.
Let us first take a quick look to the fibroblasts:
Fibroblasts treated with TGFβ differentiate into myofibroblasts (therefore they express αSMA) and collagen production is increased. At the molecular level, addition of TGFβ causes SMAD phosphorylation (and activation). So far so good.
Treating these fibroblasts with T3 (thyroid hormone) decreases SMAD activation and they end up expressing less αSMA and less collagen (the treated myofibroblast is, somehow, “lessof amyofibroblast”).
Hence, T3 alters TGFβ-induced differentiation of fibroblasts into myofibroblasts.
If thyroid hormones decrease transformation of fibroblasts into myofibroblasts and collagen production in lab conditions, they may also play a role in fibrosis disease in vivo.
In order to test this idea, two models of animal fibrosis were studied: liver fibrosis and skin fibrosis. In both cases a group of mice was having T3-supplemented drinking water and the other one drank normal water (control mice). Those mice supplemented with T3 presented hyperthyroidism (high levels of T3 in blood).
The experimental model of liver fibrosis provokes hepatic cell death and an increase of blood aspartate transaminase. The highest αSMA and collagen levels were observed in the damaged livers (they contain more myofibroblasts). Hyperthyroid mice (those who were supplemented with T3 in their drinking water) had much lower αSMA and collagen levels, in other words, thyroid hormones had a protecting role against liver fibrosis.
The experimental model of skin fibrosis causes skin to become thick, tough and scaly principally due to collagen accumulation. Once again, this kind of fibrosis goes along with higher αSMA levels (yes, myofibroblasts, one more time) and also with higher SMAD activation (TGFβ, again). Thickness of the fibrotic zone decreased in hyperthyroid mice as well as all the former biological indicators. As deduced form this results, thyroid hormones protect against skin fibrosis and can alter the TGFβ/SMAD pathway.
But, can we assume from these data that it would be advisable to increase thyroid hormone levels to treat fibrosis? No, definitely not. First of all, we must take into account that these experiments were performed using cell cultures and animal models, which cannot be directly extrapolated to humans. Also, hyperthyroidism implies other complications that should not be ignored.
So, what options do we have? If we succeed in elucidating the exact molecular mechanism underlying the alteration of the SMAD pathway by the thyroid hormones, we may be able to design drugs that specially block this step of the pathway without interfering with the rest of the hormonal functions.
The article goes even further. It accumulates evidence about the process leading to this interaction, which turns out to be actual physical contact between TR and the SMAD protein. On the other hand, using diverse mutated transcription factors, the study determines which domains are involved in this inter-pathway crosstalk. These molecular studies quantify phosphorylated proteins, gene expression, location of transcription factors in the DNA… This data compilation may be useful for designing specific drugs that enable patients to improve quality of life.
References
- E. Alonso-Merino et al (2016) Thyroid hormones inhibit TGF-β signaling and attenuate fibrotic responses PNAS doi: 10.1073/pnas.1506113113 ↩
2 comments
[…] *Imagen de portada: Estructura cristalina de la triple hélice de colágeno (Wikimedia Commons). *El artículo completo en inglés está disponible en Mapping Ignorance. […]
[…] Check our first article in Mapping Ignorance: The mystery of collagen and thyroid hormone: a detective story. […]