Modified DNA catalysts for chemical reactions in water
Modified DNA catalysts for chemical reactions in water
We all know that the main role of DNA is the storage of genomic information leading to the biosynthesis of proteins via diverse forms of RNA. In turn, proteins play multiple roles in living systems, catalysis being among the most important ones. These are the standard functions that we may find in any general chemistry textbook.
But recent discoveries add more to the complexity of these molecules: they are also catalysts. This means that they can increase the rate of a chemical reaction without themselves undergoing any permanent chemical change, providing an alternative pathway by which the reaction can proceed as it has a lower activation energy. Catalytic RNAs are called ribozymes and DNAs, deoxyribozymes, because they act in a way similar to enzymes. Actually, the catalytic activity of RNA is an important element in theories of the origin of life, particularly the RNA world concept.
In the case we may have the need to use DNA as a catalyst, we will find ourselves with the difficulty that it is structurally less versatile than RNA and proteins. In order to solve this, additional molecules and functional groups would be required to expand the catalytic space of DNA. The feasibility of this concept by intercalation of aza-chalcones in the double helix and subsequent coordination to copper salts was brilliantly demonstrated by Roelfes and Feringa in 2005.
Further work demonstrated that various intercalating heterocycles and metallic salts in the presence of DNA are able to promote, among other transformations, Diels–Alder reactions, Friedel–Crafts alkylations and Michael (including oxa-Michael) additions. It is remarkable that all these reactions were carried out in water, although in several cases the system tolerated organic co-solvents.
Despite the relevance of (3 + 2) cycloadditions in the chemical synthesis of five-membered rings, the enzymatic version of this reaction has not been identified in living systems. Only a very recent example of a possible enzymatic 1,3-dipolar reaction has been reported to date. In addition, nonenzymatic 1,3-dipolar reactions have been postulated in the biosynthesis of several alkaloids and natural products.
Now, a team of researchers from UPV/EHU and DIPC, has described 1 the first example of a DNA-assisted 1,3-dipolar reaction in water. It is a biomolecule-assisted (3 + 2) cycloaddition between azomethine ylides and alkenes to produce unnatural proline derivatives.
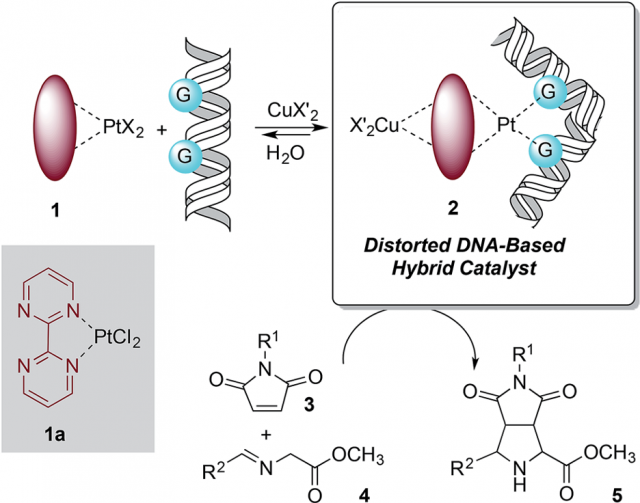
The design of the covalent modification of DNA was based on the ability of Pt(II) chemotherapeutic drugs to bind mainly 1,2-intrastrand GpG units, i.e., regions of DNA where a guanine nucleotide is followed by another guanine nucleotide (GpG is short hand for guanine-phosphate-guanine), thus providing a concave-convex distortion of the double helix that could mimic the active sites of metalloenzymes. In a previous work the researchers had reported that new chiral ligands can bind Cu(II) salts and efficiently catalyze (3 + 2) cycloadditions involving azomethine ylides. They reasoned that a DNA–Pt(II)–Cu(II) heterobimetallic complex similar to that depicted in Figure 1 could catalyze this reaction. Something that was feasible according to computational-modelling calculations.
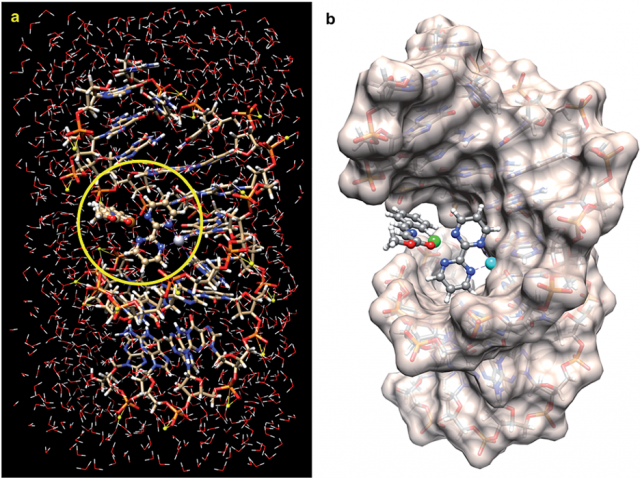
The results indicate that a suitable bimetallic complex based on Pt(II) can distort the double helix of DNA to generate an active site similar to those found in well-known metalloenzymes. The proposed reaction mechanism, based on quantum mechanical calculations, is compatible with thermally allowed mechanisms leading to the exclusive formation of racemic endo-cycloadducts.
This work demonstrates that modified biomolecules can catalyse chemical reactions in water, for which there is no equivalent in living systems.
Author: César Tomé López is a science writer and the editor of Mapping Ignorance.
References
- Iván Rivilla, Abel de Cózar, Thomas Schäfer, Frank J. Hernandez, Alexander M. Bittner, Aitziber Eleta-Lopez, Ali Aboudzadeh, José I. Santos, José I. Miranda and Fernando P. Cossío (2017) Catalysis of a 1,3-dipolar reaction by distorted DNA incorporating a heterobimetallic platinum(II) and copper(II) complex Chemical Science doi: 10.1039/c7sc02311a ↩