Chromatic multiphoton serial microscopy can generate brain-wide atlas-like colour datasets with subcellular resolution
Chromatic multiphoton serial microscopy can generate brain-wide atlas-like colour datasets with subcellular resolution
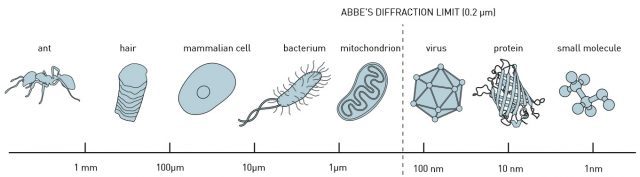
In 1873, the microscopist Ernst Abbe stipulated a physical limit for the maximum resolution of traditional optical microscopy: 0.2 micrometers, or 200 nanometers (the shortest wavelength for visible light, the extreme limit of violet). This meant that scientists could distinguish whole cells, as well as some parts of the cell called organelles. However, they would never be able to discern something as small as a normal-sized virus or single proteins.
Abbe’s equation still holds but has been bypassed just the same. Eric Betzig, Stefan W. Hell and William E. Moerner were awarded the Nobel Prize in Chemistry 2014 for having taken optical microscopy into a new dimension using fluorescent molecules. Theoretically there is no longer any structure too small to be studied. As a result, microscopy has become nanoscopy.
In 1997, Moerner demonstrated that it is possible to optically control fluorescence of single molecules. This solved a problem that Eric Betzig had formulated two years earlier. Betzig had already detected fluorescence in single molecules using near-field microscopy. He began to ponder whether a regular microscope could yield the same high resolution if different molecules glowed with different colours, such as red, yellow and green. The idea was to have the microscope register one image per colour. If all molecules of one colour were dispersed and never closer to each other than the 0.2 micrometres stipulated by Abbe’s diffraction limit, their position could be determined very precisely. Next, when these images were superimposed, the complete image would get a resolution far better than Abbe’s diffraction limit, and red, yellow and green molecules would be distinguishable even if their distance was just a few nanometres. In this manner Abbe’s diffraction limit could be circumvented. However, there were some practical problems, for instance a lack of molecules with a sufficient amount of distinguishable optical properties. 1
The real breakthrough came in 2005, when he stumbled across fluorescent proteins that could be activated at will, similar to those that W. E. Moerner had detected in 1997 at the level of a single molecule. Betzig realized that such a protein was the tool required to implement the idea that had come to him ten years earlier. The fluorescent molecules did not have to be of different colours, they could just as well fluoresce at different times.
Today, multicolour fluorescence microscopy has become a key enabling technology in biology as it provides the means to spectrally resolve cells, organelles, or molecules within tissues and to analyze their interactions. Strategies combining multiple distinct fluorescent labels are increasingly used to study spatial relationships among cells and molecules and to encode parameters such as neuronal connectivity, cell lineage, cycling state, subtype identity, genotype, or signaling pathway activation. However, scaling up such approaches at the whole organ or tissue level would be a major step forward but the lack of suitable large-volume multicolour microscopy methods is the major obstacle.
In recent years, serial block-face imaging, which relies on the automated, iterative alternation of imaging and microtome-based sectioning of a block of tissue, has been successfully transposed from electron microscopy to light microscopy. This scheme has emerged as an effective means for generating data encompassing mm3-to-cm3 volumes of tissue at subcellular-resolution with either discrete or continuous sampling. One particularly active field of application is neuroscience, where block-face fluorescence imaging has opened the way to brain-wide mesoscale connectomics and single neuron reconstruction efforts.
But to image multiplexed or combinatorial fluorescent signals with micrometer-scale precision in samples exceeding a few hundreds of microns in depth using serial block-face imaging faces serious problems. Dual-colour limitations, chromatic aberrations and channel registration over large volumes are the main ones.
Now, an international team of researchers that includes Ikerbasque Research Fellow (UPV/EHU & DIPC) Ignacio Arganda-Carreras has developed a method 2 for volume multicolour and multi-contrast microscopy with submicrometer registration of the image channels. The new approach, termed chromatic multiphoton serial (ChroMS) microscopy, relies on the integration of trichromatic two-photon excitation by wavelength mixing with automated serial tissue sectioning. The performance of ChroMS microscopy is tested for tridimensional imaging of mouse brains labeled by transgenic, electroporation-based or viral multicolour approaches.

Although clearing techniques (e.g. CLARITY, CUBIC, or iDISCO) combined with light-sheet microscopy have constituted major breakthroughs in large-volume and organ-scale tissue imaging with single-cell resolution, none of the plane illumination techniques available thus far have been able to deliver multicolour images with high contrast in all channels, together with consistent and homogeneous submicron resolution across large volumes. ChroMS microscopy achieves efficient one-shot excitation in three distinct spectral bands, enabling multicolour imaging of minimally processed tissue with sub-micron channel alignment and resolution preserved over cubic millimeter volumes. It solves two general problems encountered in tissue imaging: chromatic aberrations (inherent to all multicolour microscopy approaches) and inhomogeneous image quality across volumes.
In short, ChroMS microscopy can generate brain-wide atlas-like colour datasets with subcellular resolution as well as continuous 3D datasets of supra-millimetric size with submicron multicolour precision and diffraction-limited axial resolution.
The authors note one limitation of the ChroMS system though: the image acquisition time, as imaging an entire mouse brain with discrete axial sampling currently takes 2–3 days. This caveat, however, is neither unbreakable nor truly limiting: imaging speed in a point-scanning approach is essentially limited by the fluorescence flux, which sets the minimal pixel dwell time.
Importantly, there is no reason for all applications illustrated in the brain to be applied to organ-scale studies in other tissues. ChroMS will therefore be an asset for multiscale and system-level studies in neuroscience and beyond.
Author: César Tomé López is a science writer and the editor of Mapping Ignorance.
References
- How the optical microscope became a nanoscope. NobelPrize.org. Nobel Media AB 2019. Thu. 25 Apr 2019. ↩
- Lamiae Abdeladim, Katherine S. Matho, Solène Clavreul, Pierre Mahou, Jean-Marc Sintes, Xavier Solinas, Ignacio Arganda-Carreras, Stephen G. Turney, Jeff W. Lichtman, Anatole Chessel, Alexis-Pierre Bemelmans, Karine Loulier, Willy Supatto, Jean Livet & Emmanuel Beaurepaire (2019) Multicolor multiscale brain imaging with chromatic multiphoton serial microscopy Nature Communications doi: 10.1038/s41467-019-09552-9 ↩