CXCR4 as a therapeutic target in cancer
CXCR4 as a therapeutic target in cancer
Author: Marta Irigoyen is a postdoctoral researcher at CIC bioGUNE
In a paper led by Passaro and coworkers we sought to identify a functional link between the activity of a protein called calcineurin and the response of a type of leukemic cells known as “T-ALL” to microenvironmental signals. Furthermore, we wanted to determine whether interrupting these signals could affect to leukemia development, thus providing alternative strategies for current T-ALL therapy 1.
T-ALL is an aggressive malignancy which results from the leukemic transformation of T-cell progenitors (the cells responsible to generate the white blood cells called lymphocytes T) into tumor cells. Despite the tremendous progress in T-ALL therapies during the last decades, an elevate percentage of patients shows resistance and the fearsome tumor relapse. Leukemia initiating cells (abbreviated as LIC) are thought to be the T-ALL-initiating/propagating cells responsible for relapse following chemotherapy 2. LICs are a small subpopulation of cells within leukemic cells with capabilities of self-renewal and differentiation (such as the stem cells) responsible to provoke leukemia (re)generation. For this reason, these cells are hypothesized to resist current therapies and cause relapse and metastasis by giving rise to new tumors. Combining the experience of other types of leukemias, the researchers reached to the conclusion that LICs resistance to treatment probably involves both deregulation of molecular pathways and response to microenvironmental signals.
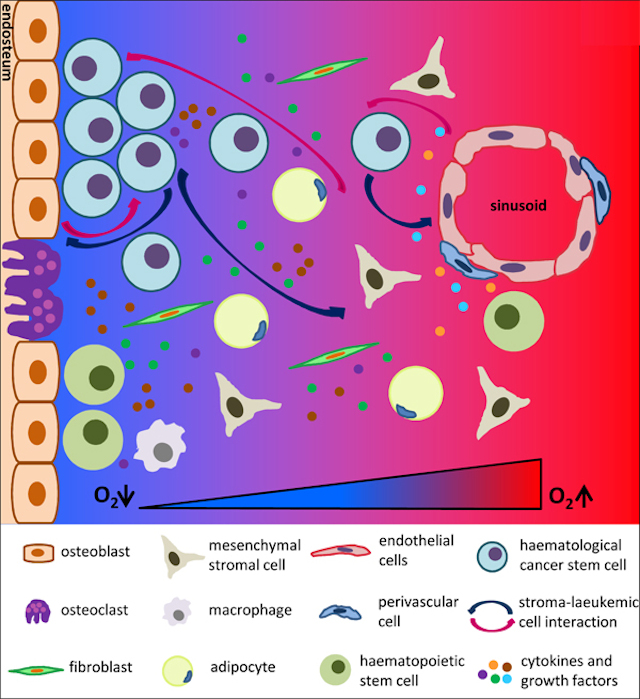
One of the triggering signals is driven by the protein calcineurin. It has been shown to be activated in T-ALL 3 and identified as essential to LIC activity in T-ALL mouse models 4. Calcineurin regulates the interaction that T-ALL cells established with other cells present in the tumor microenvironment. Notably, calcineurin regulates LICs adhesive and migratory properties promoting that these cells “escape” from their niches (the “niche” is the in vivo microenvironment where stem cells reside and receive stimuli that determine their fate) disseminating and spreading through the body. We can observed in figure 1 how LIC can establish interaction with different cell types and respond to several growth factors and cytokines presented in the niche.
To identify calcineurin-dependent regulators of T-ALL cell migration, we planned an experiment with the gain or the loss of Calcinceurin funcion. We compared the motility of calcineurin-competent and calcineurin-deficient leukemic cells in response to several signal messengers that use the white blood cells called “chemokines” and “cytokines”. We found that the chemokine “CXCL12” strongly induced T-ALL cell motility. Interestingly, we showed that calcineurin deficiency (thanks to the experiment of loss of function) impaired CXCL12-induced T-ALL cell motility. This phenotype was accompanied by a significant reduction of the chemokine receptor “CXCR4” on the cell-surface. Interestingly, this reduction on the cell surface was not accompanied to a mRNA (the messenger with instructions to fabricate the receptor) or internal levels of the receptor. To really known what was happening we must to take into consideration that the “steady-state” of CXCR4 cell-surface expression results from a balance between endocytosis (that is when the receptor is internalized into the cell), intracellular trafficking (the movement inside the cell), and recycling (when a receptor that has been internalized is re-used is conduced again to emerge in the cell membrane) 5 6. To identify the calcineurin-dependent steps in CXCR4 dynamics, we compared CXCR4 endocytosis and recycling between calcineurin-competent and calcineurin-deficient T-ALL cells. Thus, we observed no significant differences between calcineurin-competent and calcineurin-deficient T-ALL cells for CXCR4 internalization in response to CXCL12 but we observed a severe delay in CXCR4 recycling in calcineurin-deficient cells. To directly link the motility defect seen in calcineurin-deficient T-ALL cells to reduction of CXCR4 surface expression, we planned an experiment trying to restore normal cell-surface expression levels of CXCR4 in calcineurin-deficient leukemic cells. Interestingly, restoring surface CXCR4 expression in calcineurin-deficient cells to that of calcineurin-competent cells, rescued the motility defect of calcineurin-deficient cells and corrected their migratory defect.
To gain insight into the molecular mechanism underlying defective CXCR4 migration in calcineurin-deficient leukemic cells, we analyzed the calcineurin-dependent transcriptome (this is the set of all RNA transcripts presented in a cell, that means, all the “instructions” that are ongoing in the cell in a precise moment) for T-ALL cells (4). We focused our search in the deregulation of genes involved in receptor recycling. Interestingly, comparing calcineurin-deficient respect to calcineurin-competent T-ALL cells, we found a downregulation (reduction of the expression) of a protein called “cortactin”. Cortactin is an actin-binding protein involved in cytoskeleton dynamic processes (necessary for the above-mentioned intracellular trafficking). Cortactin has been reported to regulate CXCL12 response of epithelial cells overexpressing CXCR4 7 8. Thus, we investigated whether reduction of surface CXCR4 expression in calcineurin-deficient T-ALL cells was a consequence of cortactin downregulation. To do that, we generated calcineurin-competent and calcineurin-deficient T-ALL cells overexpressing exogenous cortactin. Thus, we compared cell-surface expression of CXCR4 and the migratory properties of the cells. Strikingly, we found that the induction of cortactin expression in calcineurin deficient cells was able to restore CXCR4 surface expression and correct its migratory defect to levels similar to those of calcineurin-competent cells.
To further investigate the importance of CXCR4 in T-ALL we silenced CXCR4 expression in T-ALL cells. We found that CXCR4 silencing inhibited CXCL12-induced motility and migratory properties of T-ALL cells. Moreover, we observed that CXCR4 silencing also inhibited the expansion of leukemic cells, due to increased cell death and altered cell-cycle progression. We decided to analyze the effect of CXCR4 silencing of T-ALL cells on its in vivo behavior grafting them into mice. We focused the study to the cell-“homing” (the ability of leukemic cells to reach the bone marrow) and the LIC activity (the ability of leukemic cells to generate the disease). After 24h we observed a significant reduction of homed leukemic cells in CXCR4-silenced T-ALL cells as compared to control. We also noticed a decreased viability of CXCR4-silenced leukemic cells in the bone marrow compared to control. Next, we analyzed the LIC potential of both CXCR4-silenced and control leukemic comparing leukemia development in these mice. As expected, control leukemic cells induced a fatal leukemia in all animals. However, internal dissemination and spreading of the leukemic CXCR4-silenced cells was abrogated in the 33% of mice and delayed in all the remaining animals.
To extend these findings to human T-ALL, we first silenced CXCR4 in a cell line of human T-ALL cells (Jurkat) and analyzed their in vitro and in vivo phenotypes. We found that CXCR4 silencing inhibited Jurkat cell migration and proliferation while increasing cell death in vitro and impaired the ability of Jurkat cells to generate leukemia in vivo. To further investigate CXCR4 importance in human T-ALL, we used a patient-derived T-ALL cells called “M106” 9. Similarly, whereas control leukemic cells gradually accumulated in the blood and bone marrow of recipient mice, CXCR4 silencing severely inhibited leukemia engraftment (the ability to reach the bone marrow and generate the disease). All these changes translated into an increased survival of mice injected with CXCR4-silenced T-ALL cells as compared to control.
In conclusion, in this paper we demonstrated for the first time that CXCR4 plays a fundamental role in T-ALL survival, proliferation and migration both in vitro and in vivo engraftment and leukemogenicity. Thus, these results pave the way for future clinical trials using CXCR4 inhibitors as a single treatment or in combination with current therapies to prevent T-ALL relapse.
References
- Passaro D, Irigoyen M, Catherinet C, Gachet S, Da Costa De Jesus C, Lasgi C, Tran Quang C, Ghysdael G. CXCR4 Is Required for Leukemia-Initiating Cell Activity in T Cell Acute Lymphoblastic Leukemia. Cancer Cell. 2015; 27: 769-79. PMID: 26058076 DOI: 10.1016/j.ccell.2015.05.003 ↩
- Clappier E, Gerby B, Sigaux F, Delord M, Touzri F, Hernandez L, Ballerini P, Baruchel A, Pflumio F, Soulier J. Clonal selection in xenografted human T-cell acute lymphoblastic leukemia recapitulates gain of malignancy at relapse. J. Exp. Med. 2011; 208: 653-661. PMID: 21464223 DOI: 10.1084/jem.20110105 ↩
- Medyouf H, Alcalde H, Berthier C, Guillemin MC, dos Santos NR, Janin A, Decaudin D, de Thé H, Ghysdael J. Targeting calcineurin activation as a therapeutic strategy for T-cell acute lymphoblastic leukemia. Nat. Med. 2007; 13: 736-741. PMID: 17515895 DOI: 10.1038/nm1588 ↩
- Gachet S, Genescà E, Passaro D, Irigoyen M, Alcalde H, Clémenson C, Poglio S, Pflumio F, Janin A, Lasgi C et al. Leukemia-initiating cell activity requires calcineurin in T-cell acute lymphoblastic leukemia. Leukemia. 2013; 27: 2289-2300. PMID: 23689515 DOI: 10.1038/leu.2013.156 ↩
- Grundler R, Brault L, Gasser C, Bullock AN, Dechow T, Woetzel S, Pogacic V, Villa A, Ehret S, Berridge G et al. Dissection of PIM serine/threonine kinases in FLT3-ITD-induced leukemogenesis reveals PIM1 as regulator of CXCL12-CXCR4-mediated homing and migration. J. Exp. Med. 2009; 206: 1957-1970. PMID: 19687226 DOI: 10.1084/jem.20082074 ↩
- Masztalerz A, Zeelenberg IS, Wijnands YM, de Bruijn R, Drager AM, Janssen H, Roos E. Synaptotagmin 3 deficiency in T-cells impairs recycling of the chemokine receptor CXCR4 and thereby inhibits CXCL12 chemokine-induced migration. J. Cell Sci. 2007; 120: 219-228. PMID: 17179206 DOI: 10.1242/jcs.03328 ↩
- Cosen-Binker .I, Kapus A. Cortactin: the gray eminence of the cytoskeleton. Physiology (Bethesda). 2006; 21: 352-361. PMID: 16990456 DOI: 10.1152/physiol.00012.2006 ↩
- Luo C, Pan H, Mines M, Watson K, Zhang J, Fan GH. CXCL12 induces tyrosine phosphorylation of cortactin, which plays a role in CXC chemokine receptor 4-mediated extracellular signal-regulated kinase activation and chemotaxis. J. Biol. Chem. 2006; 281: 30081-30093. PMID: 16905744 DOI: 10.1074/jbc.M605837200 ↩
- Uzan B, Poglio S, Gerby B, Wu CL, Gross J, Armstrong F, Calvo J, Cahu X, Deswarte C, Dumont F et al. Interleukin-18 produced by bone marrow-derived stromal cells supports T-cell acute leukaemia progression. EMBO Mol. Med. 2014; 6: 821-834. PMID: 24778454 DOI: 10.1002/emmm.201303286 ↩