Catching intramolecular vibrational redistribution in real time
Catching intramolecular vibrational redistribution in real time
Molecules are never truly still. Even in apparently stable matter, atoms vibrate continuously, stretching and bending the chemical bonds that hold them together. These vibrations are not random noise: they determine how molecules absorb light, exchange energy, and undergo chemical reactions. One of the central challenges in chemistry is learning how to direct energy into a specific molecular vibration in order to steer a reaction along a desired pathway. The difficulty is that molecules rapidly redistribute this energy internally, typically within a few trillionths of a second — too fast for most techniques to track.
This ultrafast process is known as intramolecular vibrational redistribution. When one vibrational mode is excited, its energy spreads into other vibrations through subtle couplings that arise from the anharmonic nature of molecular motion. Real molecules do not behave like perfectly independent springs. Their vibrations interact, allowing energy to travel through the molecule. Understanding these pathways is essential for any attempt to steer chemistry at the molecular scale.
New research 1 explores whether intramolecular vibrational redistribution can be detected in a single molecule using highly sensitive optical techniques based on surface-enhanced Raman spectroscopy, or SERS. Raman spectroscopy works by shining laser light on a molecule and measuring the frequency shifts that appear when photons exchange energy with molecular vibrations. Normally these signals are extremely weak, but metallic nanostructures can concentrate electromagnetic fields — through a quantum optical phenomenon called plasmon resonance — into nanoscale regions and amplify the interaction enormously. In carefully engineered structures, the signal becomes strong enough to detect a single molecule.
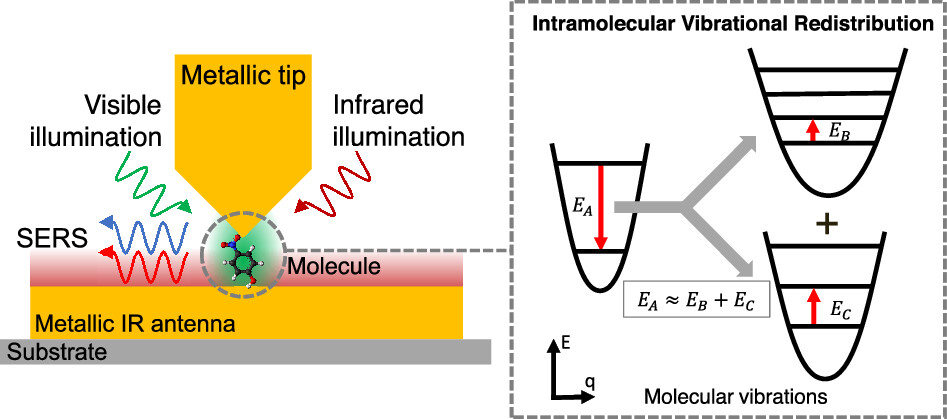
The study places a molecule inside a plasmonic nanocavity: a tiny gap, roughly one nanometer wide, between metallic structures that trap light very efficiently. The concentrated electromagnetic field inside this gap strongly enhances Raman scattering. The theoretical model combines quantum optics with molecular vibration theory to describe how light, cavity modes, and molecular vibrations interact simultaneously.
A molecular coupling with a long history
Particular attention is given to a mechanism called a Fermi resonance. This occurs when the energy of one vibrational mode nearly matches the combined energy of two quanta of another vibration, causing the two states to become strongly coupled. Rather than behaving independently, they mix into hybrid states that can exchange energy efficiently. The phenomenon is named after physicist Enrico Fermi, who first explained it in 1931 to account for unexpected doublings in the spectrum of carbon dioxide.
One signature of this coupling can appear directly in Raman spectra. What would otherwise be a single spectral peak can split into two nearby peaks, forming what is known as a Fermi doublet. However, in realistic molecules the splitting is often small and difficult to distinguish from coincidental overlaps with other spectral features. The work therefore investigates whether the time evolution of vibrational populations offers clearer evidence of intramolecular vibrational redistribution.
Two strategies to catch energy in motion
Two pump-and-probe strategies are analyzed. In the first, visible laser pulses excite molecular vibrations through Raman scattering itself. Stokes Raman processes deposit energy into a vibrational mode, while anti-Stokes Raman scattering — which requires the molecule to already be vibrationally excited — reveals how the vibrational populations evolve over time. The calculations show that when one vibration is pumped strongly, energy flows into a second coupled vibration through the Fermi resonance, producing a measurable increase in the anti-Stokes signal associated with that second mode.
Under pulsed excitation, the dynamics become especially revealing. After the laser pulse excites one vibration, the energy oscillates back and forth between the two coupled vibrational states before eventually dissipating. These oscillations closely resemble Rabi oscillations, a well-known quantum mechanical phenomenon in which two coupled quantum systems coherently exchange energy. In the molecule studied, the predicted oscillation period is about 1.5 trillionths of a second — a timescale that falls within the detectable range of current experiments.
Another important signature is a delayed buildup of population in the secondary vibration. The first vibration is excited almost immediately by the laser pulse, but the second gains significant population only after energy has been transferred through the Fermi resonance pathway. This delay, lasting several hundred trillionths of a trillionth of a second (hundreds of femtoseconds), provides direct evidence that the second mode is populated through intramolecular vibrational redistribution rather than by direct optical excitation.
The study also investigates a second approach using infrared pumping. In this configuration, an infrared cavity mode resonantly drives a specific molecular vibration directly, while visible-light Raman scattering simultaneously probes the resulting populations. Infrared excitation drives the selected vibration into a coherent quantum state, producing a sharp and intense anti-Stokes signal. At higher pumping strengths, population transfer through the Fermi resonance again enhances the Raman signal of the coupled vibration, revealing the underlying intramolecular vibrational redistribution pathway with a clarity that pure spectral inspection cannot provide.
Within reach of experiments
The calculations use realistic parameters for gold and silver plasmonic nanocavities and for a molecule commonly studied in SERS experiments: 4-nitrobenzenethiol. The predicted coupling strengths, vibrational lifetimes, cavity losses, and field enhancements are all consistent with values reported in current experiments. The work therefore suggests that observing intramolecular vibrational redistribution at the single-molecule level may already be within reach of existing nanophotonic platforms, particularly when operated at cryogenic or near-cryogenic temperatures where thermal noise is reduced.
Beyond spectroscopy, the results point toward a broader ambition: controlling how energy moves through molecules. If individual vibrational pathways can be monitored and eventually manipulated, it may become possible to direct chemical reactions with unprecedented precision. Nanophotonic cavities could then act not only as ultrasensitive detectors, but also as tools for engineering molecular dynamics themselves — turning what was once an invisible and uncontrollable process into something that can be seen, studied, and ultimately steered.
Author: César Tomé López is a science writer and the editor of Mapping Ignorance
Disclaimer: Parts of this article may have been copied verbatim or almost verbatim from the referenced research paper/s.
References
- Aurelian Loirette-Pelous, Roberto A. Boto, Javier Aizpurua, and Ruben Esteban (2026) Addressing Intramolecular Vibrational Redistribution in a Single Molecule through Pump and Probe Surface-Enhanced Vibrational Spectroscopy ACS Photonics doi: 10.1021/acsphotonics.6c00030 ↩