Overcoming bottlenecks in bio-interface simulations: The MartiniSurf Approach
Overcoming bottlenecks in bio-interface simulations: The MartiniSurf Approach
Enzymes and other biomolecules are often anchored onto solid materials so they can be reused, purified more easily, and made more stable, a strategy biotechnology has relied on since industrial-scale immobilized enzymes first appeared in chemical manufacturing in the mid-twentieth century. Immobilized enzymes today drive processes from food and pharmaceutical production to biosensors and medical diagnostics. Fixing a biomolecule in place, however, is not as simple as gluing it down. The exact spot where it attaches, the direction it faces, and how much freedom it retains to move can all change how well it works. Testing every possible arrangement in the laboratory is slow and costly, which is why computer simulation has become such a valuable complement to experiments.
The Martini force field
One of the main tools for this kind of study is molecular dynamics simulation, which calculates how atoms move over time by applying the basic laws of physics. Simulating every single atom of a protein, its surrounding water, and a supporting material gives rich detail, but is extremely demanding on computing power, especially when many attachment geometries need comparing. A widely used shortcut is coarse-grained modeling, in which small clusters of atoms are grouped into single, simplified particles. This keeps the essential physical behavior of the system while making the calculations far cheaper. The most established coarse-grained approach for biological molecules is the Martini force field, first developed in 2004 to describe fatty, membrane-forming molecules and later extended to proteins and, more recently, to a more accurate third generation of the model. Martini has since become a standard tool for simulating large biological assemblies that would otherwise be out of reach.
Introducing MartiniSurf
A team of researchers now introduces MartiniSurf 1, an open-source software package that automates the preparation of these coarse-grained simulations for biomolecules attached to surfaces. Building such a system by hand normally involves several separate programs and many manual steps, which makes it hard to reproduce results or compare different setups fairly. MartiniSurf instead combines the whole process into a single, streamlined workflow. Starting from the structure of a protein or a DNA strand, it converts the molecule into its coarse-grained form, builds a model of the supporting surface, positions the molecule in a chosen orientation, adds water and dissolved ions, and produces all the files needed to run the simulation with GROMACS, a molecular dynamics program that has been developed and refined for decades. Because every step is controlled by explicit settings rather than manual adjustments, two researchers using the same settings should obtain the same starting system, making comparisons across studies far more reliable.
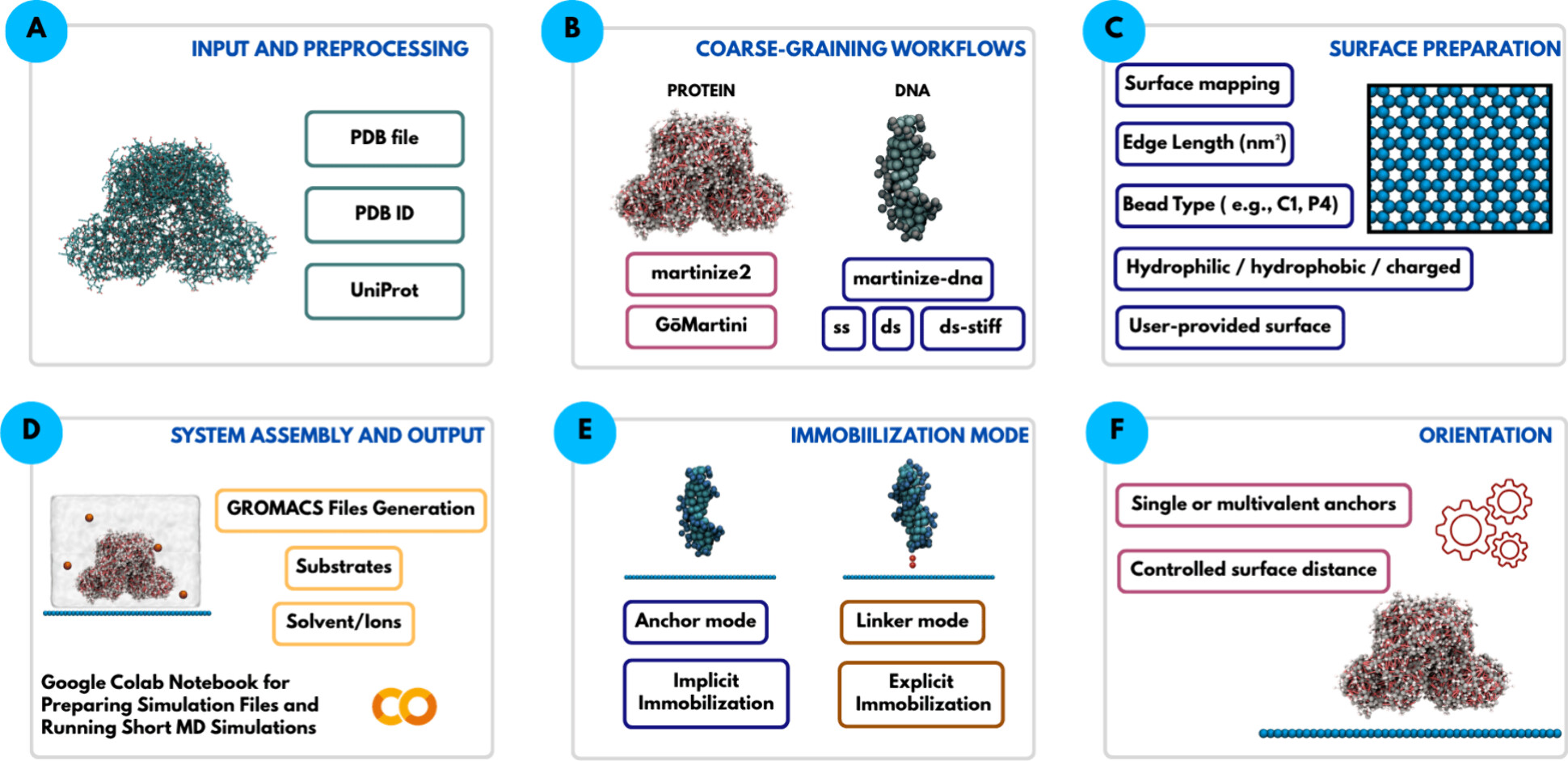
Restraining forces or linkers
A central feature of the software is its flexibility in representing how a biomolecule is attached. In one mode, selected regions of the molecule are held near the surface through simple restraining forces, without modeling the physical connector itself. In another mode, an explicit molecular linker is placed between the biomolecule and the surface, so that the linker’s own length and flexibility can influence the simulation. Because the properties of such linkers are known to affect how active and stable an attached enzyme remains, this second option offers a more realistic picture whenever the necessary coarse-grained description of the linker is available.
Variety of supporting surfaces
The supporting surfaces themselves can also be varied widely. The software can generate simple flat reference surfaces or draw on previously developed coarse-grained models of real materials such as graphene, graphite, carbon nanotubes, and agarose-like gels, the type of porous material commonly used to immobilize enzymes in the laboratory. Properties such as surface chemistry, electric charge, particle spacing, and added chemical groups can all be adjusted. Recent studies have shown that surfaces built without care can cause water molecules near the interface to line up in unrealistic, almost frozen patterns, an artifact rather than genuine physical behavior. The software includes options that let users introduce controlled irregularity into the surface to reduce this problem, while staying consistent with the wider Martini modeling framework.
In practice
To demonstrate how the approach works in practice, the study presents examples involving both proteins and DNA. For an enzyme attached to an agarose-like surface, the software reproduces several different attachment arrangements, including a complete system containing the enzyme together with its natural helper molecule and reaction substrate. These examples show how changing the attachment point or adding chemical groups to the surface can be explored systematically, without needing to reconstruct every model from scratch by hand.
The approach is also extended to DNA tethered to a graphene-like surface, comparing an electrically neutral surface with a positively charged one. The coarse-grained simulations reproduce the same general behavior previously reported in much more computationally demanding all-atom studies: the DNA strand remains mostly upright above the neutral surface but bends progressively toward the surface when it carries a positive charge, drawn in by electrostatic attraction. This comparison is not meant to reproduce exact experimental values, but rather to confirm that the automated setup captures the right physical trends, providing a check on the reliability of the workflow.
Reproducibility
Every simulation generated by the software automatically records the settings, software versions, and modeling choices used to build it, so that another researcher can repeat or directly compare a given calculation later. Importantly, the software does not introduce new physical models or new force-field parameters of its own. Instead, it draws on already validated Martini tools and organizes them into a consistent, automated pipeline.
Overall, MartiniSurf addresses a practical bottleneck in computational chemistry rather than proposing a new simulation method. By automating the construction of surface-bound protein and DNA systems, it makes coarse-grained molecular dynamics studies faster to set up, easier to reproduce, and simpler to scale across many attachment geometries. This kind of tool should make it easier to systematically study how surface properties, attachment geometry, and molecular flexibility shape the behavior of immobilized biomolecules, supporting the design of better catalysts, biosensors, and other biotechnology applications.
Author: César Tomé López is a science writer and the editor of Mapping Ignorance
Disclaimer: Parts of this article may have been copied verbatim or almost verbatim from the referenced research paper/s.
References
- J. C. Jimenez-Garcia, F. Lopez-Gallego, X. Lopez, and D. De Sancho (2026) MartiniSurf: Automated simulations of surface-immobilized biomolecular systems with Martini J. Chem Inf. Model. doi:10.1021/acs.jcim.6c00953 ↩