Spin-permutation diabatization, an intuitive map for molecular magnetism
Spin-permutation diabatization, an intuitive map for molecular magnetism
Ever since the birth of quantum mechanics in the early twentieth century, chemists have struggled with a fundamental paradox. On one hand, Lewis dot structures and molecular drawings teach us to think of electrons as localized entities—either sitting neatly in lone pairs or shared directly between two bonding atoms. On the other hand, Schrödinger’s wave mechanics views electrons as delocalized waves, spread collectively across the vast architecture of an entire molecule. This collective view is standard in modern computational chemistry, where computer algorithms solve for what are known as adiabatic states. These states provide highly accurate energy levels, but they strip away our intuitive, real-space picture of where individual electron spins actually reside.
This loss of clarity is particularly problematic when studying molecules with unpaired electrons. Known as radicals, diradicals, or molecular magnets, these systems possess unique magnetic, optical, and reactive properties that depend entirely on how their unpaired electron spins communicate with one another. When multiple unpaired electrons inhabit a single molecule or a cluster of molecules, their spins can align in the same direction (ferromagnetic coupling) or in opposite directions (antiferromagnetic coupling). To calculate these interactions, scientists rely on simplified historical models, such as the Heisenberg spin model, which treats molecules like networks of localized bar magnets. However, bridging the gap between a giant, complex quantum wave function and this simple bar-magnet picture has traditionally required complex mathematical projections or artificial modifications to molecular orbitals.
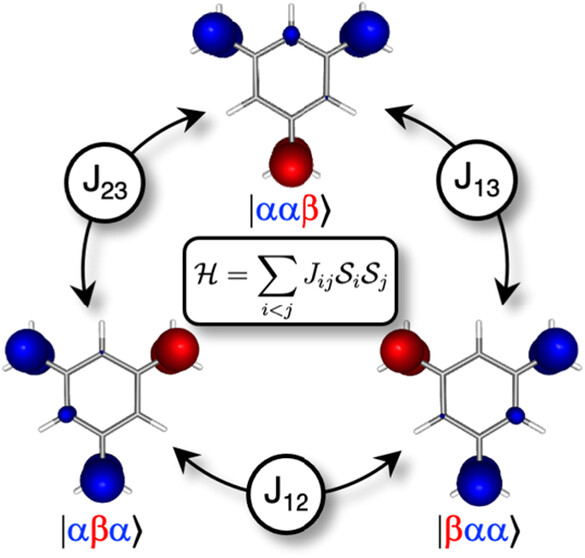
Spin-permutation diabatization
A elegant solution proposed by Alicia Omist and David Casanova 1 to this long-standing dilemma comes in the form of a framework known as spin-permutation diabatization. Instead of altering the molecular orbitals themselves, this new strategy operates directly on the spin degrees of freedom within the quantum wave function. The method utilizes a fundamental quantum mechanical tool: the spin-permutation operator, a mathematical function that interchanges the spin coordinates of two electrons.
By repeatedly applying mathematical rotations that minimize what is called self-spin exchange, the Omist-Casanova algorithm acts like an automated dealer shuffling a deck of cards. It systematically rearranges the collective, delocalized adiabatic states until they transform into “diabatic” states. In this new diabatic representation, the mathematical haze clears, revealing a vivid, real-space map of precisely where the spin-up and spin-down electron densities are localized. Because this transformation is purely unitary, it preserves the exact quantum mechanics of the system while translating it into a language that aligns perfectly with classical chemical intuition.
The power of this approach becomes obvious when observing how electron spins behave during chemical reactions or inside advanced materials. Consider the simple molecule ethylene, which consists of two carbon atoms joined by a double bond. Under normal conditions, the electrons are locked tightly in a shared bond. However, as the molecule twists around its central carbon-carbon axis, this bond progressively weakens. By tracking the molecule through this twist, the spin-permutation framework shows exactly how the collective electron cloud breaks down, revealing two distinct, localized unpaired electrons settled on opposite carbon atoms.
Solar cells and organic electronics
The Omist-Casanova strategy is also shedding light on cutting-edge materials used in solar cells and organic electronics. In organic chromophores (molecules that absorb and emit light) the energy difference between a singlet state (where electron spins are paired) and a triplet state (where spins are unpaired) dictates how efficiently a material can perform. For example, in technologies like thermally activated delayed fluorescence, which powers vivid digital displays, a tiny energy gap between these states is required. By applying spin diabatization, it becomes clear that this energy gap is dictated by spatial separation. When the calculated spin-up and spin-down densities overlap heavily in the same region of a molecule, their mutual interactions are strong, resulting in a wide energy gap. Conversely, when the spins are coaxed onto entirely different atomic sites or separate fragments of a molecular pair, the interaction weakens, and the energy gap narrows dramatically.
A translator
Ultimately, this computational breakthrough provides an elegant translator for modern chemistry. By transforming complex, collective wave functions into an intuitive landscape of localized spins, it bridges the gap between abstract quantum mathematics and practical molecular design. It offers scientists a clearer lens through which to view molecular magnets, light-harvesting systems, and strongly correlated electrons, proving that we do not have to sacrifice physical intuition to achieve quantum accuracy.
Author: César Tomé López is a science writer and the editor of Mapping Ignorance
Disclaimer: Parts of this article may have been copied verbatim or almost verbatim from the referenced research paper/s.
References
- A. Omist, and D. Casanova (2026) Spin-permutation diabatization: A general framework for spin localization and exchange coupling J. Chem. Theory Comput. doi: 10.1021/acs.jctc.5c01904 ↩