Sometimes, heavier drugs are safer
Sometimes, heavier drugs are safer
Enantiomers are any two molecules that are mirror images of each other, in the same way that, within a same person, left and right hands are: both hands look exactly the same but indeed they are not, as they cannot be superimposed. This is very easily seen when we try to put the wrong glove on: we could say that the left hand is “active” only when it comes to fit in the left glove, but not in the right one. Likewise, some molecules are active as drugs whereas their enantiomers are not. This happens because, given that most biological molecules are present in only one of their possible chiral forms, different enantiomers of a chiral drug molecule may bind differently (or not at all) to their target receptors.
The problem with this is, organic synthesis generally leads to racemates (mixtures of both enantiomers) which are very difficult to separate. Since both enantiomers often don’t have the same pharmacological properties, a single enantiomer of a drug molecule will generally be more active than the other, and obviously more active than the racemic mixture too. Besides, there is another challenge, as racemates can show limitations not only because of their lower activity but due to unwanted, harmful, or even beneficial but entirely different, unsought effects resulting from the undesired enantiomer (distomer) and/or its metabolites. For this reason, most chiral drugs are nowadays developed and sold as single enantiomers, and since the 1990s several drugs previously present in the market as racemates have been the subject of chiral switching, namely the replacement of previously developed racemic mixtures by enantiopure formulations.
Unfortunately, the development of certain chirally pure molecules is so difficult that it sometimes forces researchers to abandon valuable scaffolds in favour of alternative syntheses leading to stereochemically pure compounds. More often than desired, such syntheses are indeed achieved but then found to be hampered by racemization during storage or when the drug is metabolised. These are the reasons why, despite the dramatically improved therapeutic properties of single enantiomers over the racemic mixtures, numerous drugs and drug candidates are still developed as racemates, including the drugs derived from thalidomide.
Racemic thalidomide (Figure 1) was first commercialised in the late 1950s as a sedative, sleep-inducing drug. Research had also found that it was an effective antiemetic with a particularly good inhibitory effect on morning sickness, so it unfortunately became soon very popular among women in the first weeks of pregnancy 1. Despite the beneficial properties of the R-enantiomer, the S-enantiomer sadly became very well known after its teratogenic effects for which drugs did not have to be tested prior to being approved in the 1950s. Since it affects the limb development of the foetus, many babies were born with deformities after their mothers had taken the drug during the first three months of pregnancy, when the limbs are formed. This tragic misuse served as a lesson in drug development, revealing the need to understand the molecular pharmacology, toxicities and mechanisms of both enantiomers of a same compound.
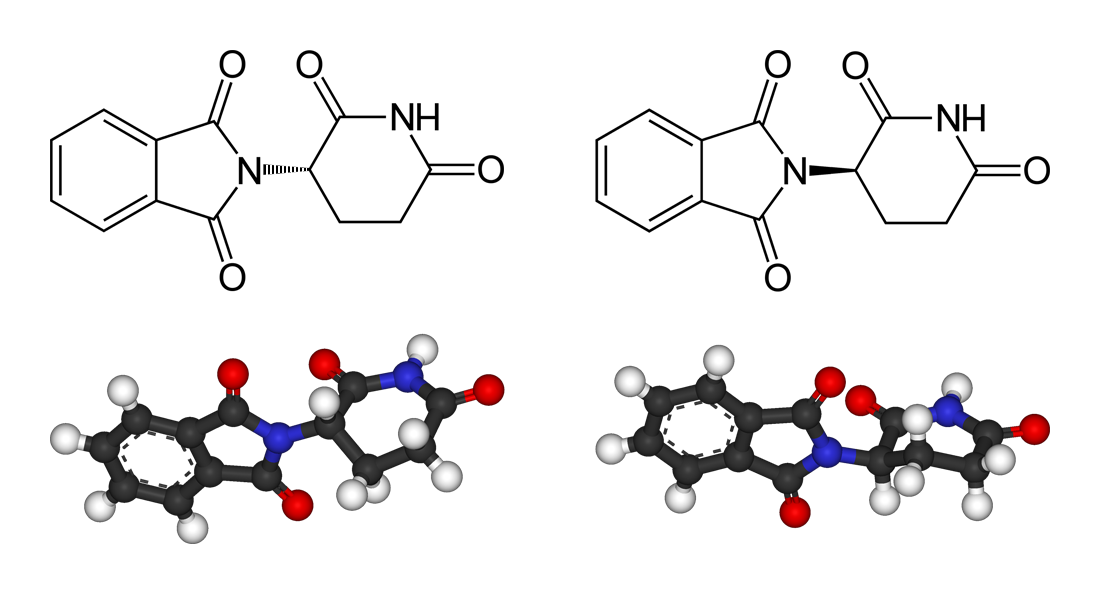
Despite its history, thalidomide and its related analogues are still interesting thanks to their antitumorigenic and anti-inflammatory properties, with applications ranging from the treatment of certain cancers to complications of leprosy and HIV infection. A chiral switch, however, cannot be applied as these drugs are known to racemize in the body at a rate faster than that of elimination, so any plan to administer a purified single enantiomer in order either to dose and study its effect, or to avoid the teratogenic effects of thalidomide, would be in vain. For that reason, the class of compounds known as immunomodulatory drugs derived from thalidomide (Figure 2) is still developed and sold as racemates nowadays.
Recently, DeWitt and coworkers 2 have resolved the racemization issue of two thalidomide analogues by replacing the acidic proton at their stereocentres with deuterium (Figure 2). The chiral centre of these molecules (the 3-aminoglutarimide moiety) is chemically unstable, resulting in rapid interconversion of the enantiomers both in vitro and in vivo. Racemization occurs by proton exchange at this stereocentre, but deuteration slows the process, while the rate of elimination of the drug from the body is not affected, and as the overall effect, elimination happens before racemization if the proton at the stereocentre is replaced by a deuterium atom.
Previous attempts of solving this issue have included replacement of the exchangeable hydrogen with methyl or fluorine groups. However, none of them were very successful or superior to the racemic, protonated thalidomide analog. On the contrary, the aforementioned substitutions were showed to lead to similar or decreased potency, increased degradation, increased toxicity, and/or increased teratogenicity. But now, replacement of the exchangeable hydrogen at the chiral centre with deuterium has allowed the stabilization and testing of individual enantiomers for two thalidomide analogues (Figure 2), CC-11006 (compound 1, Figure 2), and CC-122 (compound 2, Figure 2), which is particularly interesting as it is currently undergoing human clinical trials for haematological cancers and solid tumours.
![Figure 2. Top row: structures of thalidomide and its analogues pomalidomide and lenalidomide. The second and third rows show the structures of the compounds reported on the paper: protonated and deuterated enantiomers of compound 1 [CC-11006; i.e., (S)-H-1, (R)-H-1, (S)-D-1, and (R)-D-1] as well as protonated and deuterated enantiomers of compound 2 [CC-122; (S)-H-2, (R)-H-2, (S)-D-2, and (R)-D-2. | Credit: Czarnik et al. (2015).](https://mappingignorance.org/app/uploads/2015/05/thalidomide.jpg)
Deuterium, also known as “heavy hydrogen” since its weight is twice as much, is a stable isotope of hydrogen with a natural abundance of 0.015%, known for its potential to stabilize chemical bonds. Unlike other functional groups such as methyl or fluoro, it is predicted to not affect the pharmacological properties of a drug. Besides, given its natural abundance and its widespread use in human pharmacokinetic studies since the 1960s 3, its use in therapeutics does not present a safety concern. Therefore, the ability to stabilize and differentiate enantiomers by the “Deuterium-Enabled Chiral Switching” (DECS) described by DeWitt, not only affords a great opportunity to study and improve the activity of thalidomide analogues, but also enables the development of other drugs previously developed as racemates, thus opening a vast field in the development of safer therapeutics and the discovery of new mechanisms and clinical applications.
References
- Franks, M. E.; Macpherson, G. R.; Figg, W. D. The Lancet, 363 (9423), 2004, 1802 ↩
- Vincent Jacques, Anthony W. Czarnik, Thomas M. Judge, Lex H. T. Van der Ploeg, and Sheila H. DeWitt (2015) Differentiation of anti-inflammatory and antitumorigenic properties of stabilized enantiomers of thalidomide analogues PNAS DOI:10.1073/pnas.1417832112 ↩
- O’Driscoll, C. Chem. Ind., 2009 (5), 2009, 24 ↩