NOF approximations applied to iron(II) porphyrin
NOF approximations applied to iron(II) porphyrin
As early as the 1970s, it was suggested that one-particle reduced density matrix functional theory could be an attractive alternative formalism to wave function-based methods. Unfortunately, calculations based on exact functionals generated by the constrained-search formulation are computationally too expensive, which has prompted the development of approximate functionals for practical applications. The functionals currently in use are constructed on the basis where the one-particle reduced density matrix is diagonal, which is the definition of a natural orbital functional (NOF).
Nowadays, an open-source implementation of NOF-based methods is available to the scientific community. The associated computer program DoNOF (Donostia Natural Orbital Functional) is designed to solve the energy minimization problem of an approximate NOF, describing the ground state of an N-electron system in terms of natural orbitals and their occupation numbers. Fractional occupancies naturally allow NOFs to recover the static correlation. In fact, approximate NOFs have demonstrated to be more accurate than their electron density-dependent counterparts for highly multiconfigurational systems and scale satisfactorily compared to wave function-type methods with respect to the number of basis functions.
Recently, a NOF was proposed for electronic systems with any spin value regardless of the external potential, that is, a global NOF (GNOF). The adjective “global” is used instead of “universal” to differentiate this approximate multipurpose NOF from Valone’s exact one. GNOF is able to achieve a balanced treatment of static and dynamic electron correlations even for those systems with significant multiconfigurational character, preserving the total spin of multiplets. It should be noted that the agreement obtained by GNOF with accurate wave function-based methods is not only for relative energies but also for absolute energies, a sign of good results for good reasons.
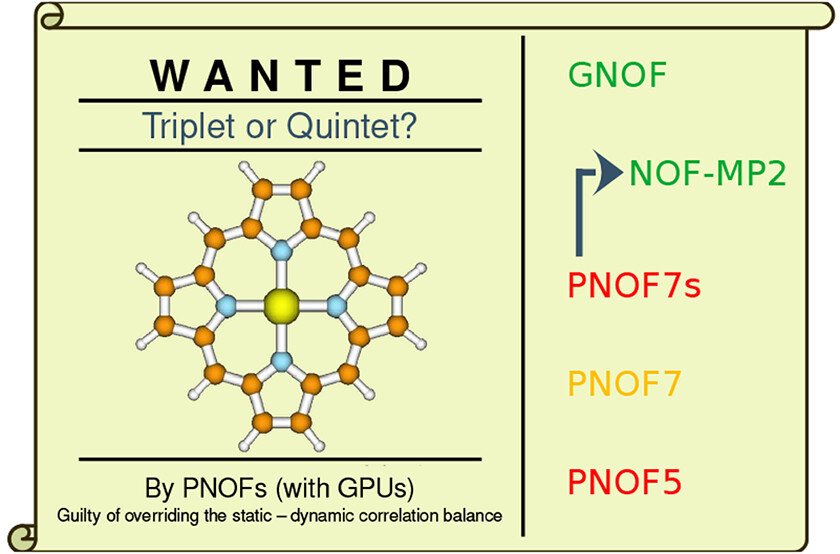
The iron porphyrins have proven to be challenging for any theoretical method and an attractive system for testing GNOF. Initial single reference studies considered a triplet state, but subsequent multireference studies favored a quintet state. The controversy about the spin of the ground state of FeP continues to this day, and the discussion has become enriched with the increase of computational power and the development of innovative methods to include electronic correlation more accurately.
Calculations with currently used single reference methods such as coupled cluster and modern density functional approximations tend to favor the triplet as the ground state, but both the triplet and quintet states have been reported to not present essential symmetry breaking. The controversy remains of active interest.
Now, a new study provides on this respect 1 important information in many ways. First, the analysis of FeP from the perspective of PNOF functionals provides information on the static and dynamic correlation effects on the problem. At the same time, it allows us to compare the set of PNOFs with the different methods previously used to study FeP. Note further that GNOF correlates all electrons into all available orbitals for a given basis set, which in the case of FeP using a double-ζ basis set correlates 186 electrons in 465 orbitals. Such a correlation calculation is not possible with current wave function-based methods.
Results show that PNOF5, PNOF7s, and PNOF7 predict the quintet to have a lower energy than the triplet state; however, the addition of dynamic correlation via second-order Møller–Plesset corrections (NOF-MP2) turns the triplet state to be lower than the quintet state, a prediction also reproduced by GNOF that incorporates much more dynamic correlation than its predecessors.
Surprisingly, there is another singlet state predicted by GNOF with a predominant dynamic correlation. In principle, this state is the one with the lowest energy, which reinforces the importance of the dynamic correlation in the stability of the iron porphyrin; however, GNOF does not contain dynamic correlation terms for the single electrons that appear in spin multiplets, so there is not a definitive answer at this time for this finding.
Author: César Tomé López is a science writer and the editor of Mapping Ignorance
Disclaimer: Parts of this article may have been copied verbatim or almost verbatim from the referenced research paper/s.
References
- Juan Felipe Huan Lew-Yee, Jorge M. del Campo, and Mario Piris (2023) Electron Correlation in the Iron(II) Porphyrin by Natural Orbital Functional Approximations J. Chem. Theory Comput. doi: 10.1021/acs.jctc.2c01093 ↩