Foreseeing the course of chemical reactions with computers and theory
Foreseeing the course of chemical reactions with computers and theory
Author: Ewa N. Szlapa is a is a Ph.D. student (ITN-EJD-TCCM) at Catholic University of Leuven
At the beginning of the development of chemistry as a science there was alchemy, which was considered to be a magicians’ business. An alchemist put some ingredients into the glass vessel and after some time the mixture inside could for example change the color. Nowadays we know that this is not magic but the fundamental blocks of all existing matter (atoms and molecules) reacting with each other and changing their properties during the course of the reaction. Furthermore the evolution of machines, computers and knowledge itself has helped people to better understand chemical reactions.
In our era of supercomputers another branch of chemistry evolved – computational chemistry. It provides an important contribution to the knowledge of chemical reactions. It gives an insight into the mechanism of transformation of one set of molecules into another. Even though its techniques and models incorporate many approximations it provides useful data about reaction species. The molecular structure of reactants (initial set of molecules that start the chemical reaction), intermediates (species that are formed from reactants but react further on to give products) and products (final molecules that are formed from the chemical reactions) together with activation energies (barriers that have to be overcome to transform chemical species) can be calculated using available quantum chemistry programs in a reasonable amount of time. The information of the structures of molecules together with the application of chemical kinetic theories can be used even to predict the course of reaction in a macroscale. A very handy and simple way to understand the kinetics (the evolution of an amount of reactants in time) is transition state theory 1. It focuses its attention on the short-lived species (transition states) positioned at the point of maximum energy connecting reactants and products. In other words if a chemical transformation is to occur the system has to possess enough energy to climb the hill represented by transition state (the idea is presented in Figure 1). Transition state is not a stable molecule and cannot be isolated from reaction mixture. However it can be located using concepts of theoretical chemistry thus providing an insight into reaction mechanism 2.
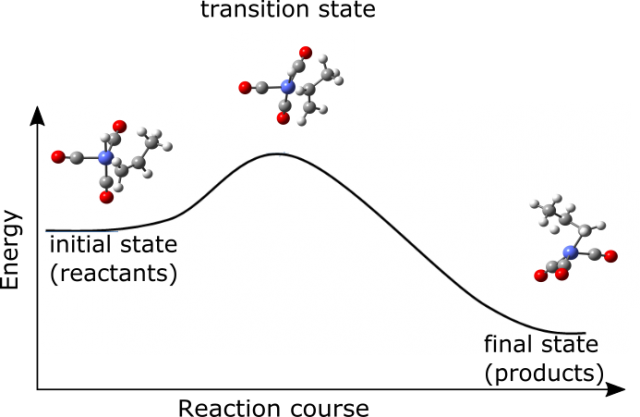
One of the ultimate goals of applied computational chemistry would be to foresee the behavior of even very complicated however highly applicable systems. An example of such a reaction, that has been extensively studied computationally, is hydroformylation 3. It is used for the transformation of cheaper reactants (in this case molecular hydrogen, carbon monoxide and alkene) into more desirable aldehydes. This reaction is already successfully commercialized. The annual world capacity of this process is eight million tones and the produced aldehydes and alcohols have a variety of applications in chemical and even pharmaceutical industry. Yet some of the features of this reaction are still not fully understood as the system is highly complicated and it is difficult to address all its aspects with scientific methods.
There are several challenges of studying hydroformylation with tools of computational and theoretical chemistry. To illustrate them we can take as an example cobalt-catalyzed propene hydroformylation (Figure 2).
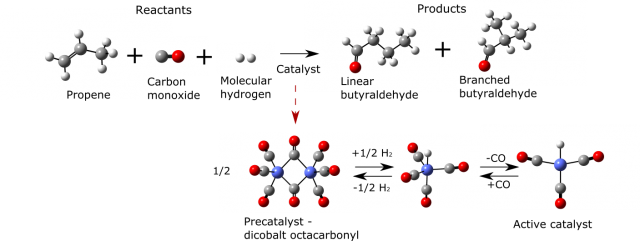
Firstly the reaction is composed of many elementary steps of which some are depicted in Figure 3. For each of them the most stable geometrical structures of reactants have to be localized in order to find the most probable way the reaction occurs. Secondly two main products appear in the reaction mixture (linear and branched butyraldehyde). These products and pathways leading to them are however highly similar in energies and the issue of selectivity (ratio of final products which is often the main concern of industry when only one is desirable for its applications) can be sometimes only addressed with expensive and long lasting calculations for which the error is significantly reduced. Later on one has to use statistical mechanics (it describes the behavior of large numbers of atoms and molecules) to account for temperature and pressure effects as well as macroscopic quantities of reacting species. Just then the rates of the reaction can be calculated using transition state theory and compared with experiment. Each step incorporates errors so the detailed picture of reaction is hard to obtain. But with a very rigorous treatment of all mentioned stages of computational studies it is starting to be possible 4.
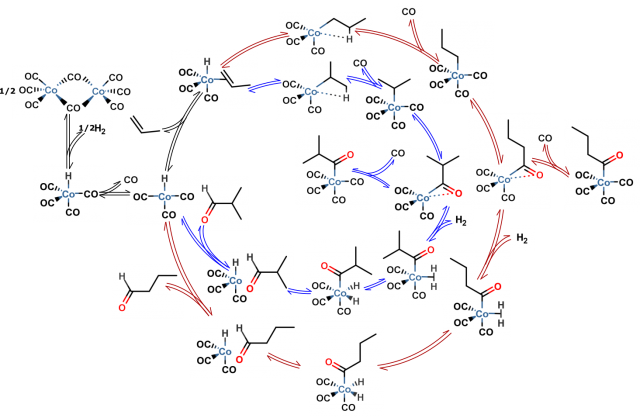
With the further development of computational techniques and computer hardware the results of theoretical chemistry studies will become even more important in the scientific field. Screening of many chemical reactions while varying catalysts, reactants, operation conditions etc. with high accuracy calculations would save time and resources needed for conducting the actual experiment and reduce the risk of working with dangerous chemicals. The experimental and theoretical work of scientists is complementary and may give foundations for designing the most efficient processes for chemical industry.
References
- K. J. Laidler, Chemical Kinetics, 3rd ed., Harper & Row, New York (1987). ISBN: 0-06-043862-2. ↩
- A. Bakac, Physical Inorganic Chemistry, Wiley, New Jersey (2010). ISBN: 978-0-047-22419-9 ↩
- T. Kégl, “Computational Aspects of Hydroformylation”, RSC Adv. 5, 4304 (2015). DOI: 10.1039/c4ra13121e ↩
- L. E. Rush, P. G. Pringle, J. N. Harvey, “Computational Kinetics of Cobalt-Catalyzed Alkene Hydroformylation”, Angew. Chem. Int. Ed. 53, 8672 (2014). DOI: 10.1002/ange.201402115 ↩
1 comment
[…] Our PhD student, Ewa Szlapa, has just published the outreach paper about her research in Mapping Ignorance. The paper is aimed at a broad public. You may have a look here: link […]