DNA replication: how a job is well done in just a few hours
DNA replication: how a job is well done in just a few hours
Think on a cell, it is alive and, among other things, it lives to reproduce itself. The paradigm of cell division is the mitosis, a wonderful dance orchestrated to equitably distribute the possessions of the mother cell: molecular structures, machinery and, especially, the genetic material. Prior to distribute, a good mother always amasses a fortune. In the interphase (the period of time in between two mitotic events) a cell usually needs to grow in size and entirely duplicate its genetic material before it is allowed to divide. Generating enough molecular structures and machinery to support the initial life of daughter cells is a big deal. But to duplicate with high fidelity the massive amount of DNA packaged in several chromosomes with the synchronicity and the strict temporal program that they do it, it is a huge and incredible task.
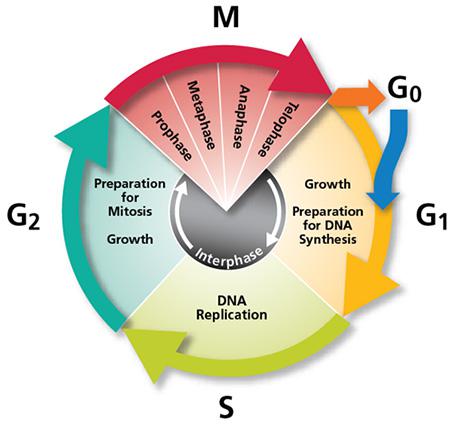
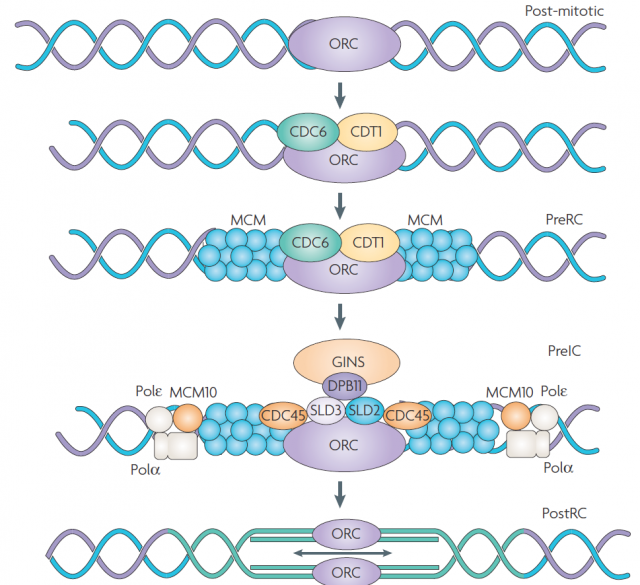
DNA replication is carried out in S phase. It begins in hundreds of replication origins distributed along each chromosome, though the preparation of this process happens much earlier at the end of the previous telophase by the assembly of pre-replicative complexes in the replication origins (Fig. 2). The sequential addition of other factors during G1-phase will be completed with DNA polymerase assembly in the replication origin at S-phase onset, starting DNA replication bidirectionally until a replication fork meets the neighbour ones. However, not all the replication origins get to begin the job; and the ones that do it do not fire at the same time. Some groups of replication origins, called “replicon clusters” because they get started together, begin replication earlier than others during S-phase. There are early and late replication clusters and the coordination of their ignition is named the replication-timing program. The replication-timing program is robust and heritable. it is known to be in disarray in many genetic illnesses, although so far we have no idea about the deep of its biological relevance, we know even less regarding the mechanisms that control so finely replication at whole genome scale1.
Since the molecular components involved in replication are quite conserved through the tree of life, some models rose trying to explain the big diversity observed in the replication ignition sequence among different organisms. This has puzzled biologists for a long time: some initiation sites only ignite in a fraction of cell cycles while others fire always but, among this stochasticity, arises a reproducible cell cycle. Further more, whereas some organisms display random initiation site recognition, others show a well-defined initiation site selection; and most metazoans are distributed in the middle of those extremes. Therefore, that is the question: How stochastic initiation events result in a whole genome replication in such an accurate, deterministic but also plastic and regulable cell cycle? Do not forget that replication happens simultaneously to the level of transcription necessary to support the live of an active cell, which is metabolically able to adapt to a changing environment and/or undergoing developmental processes.
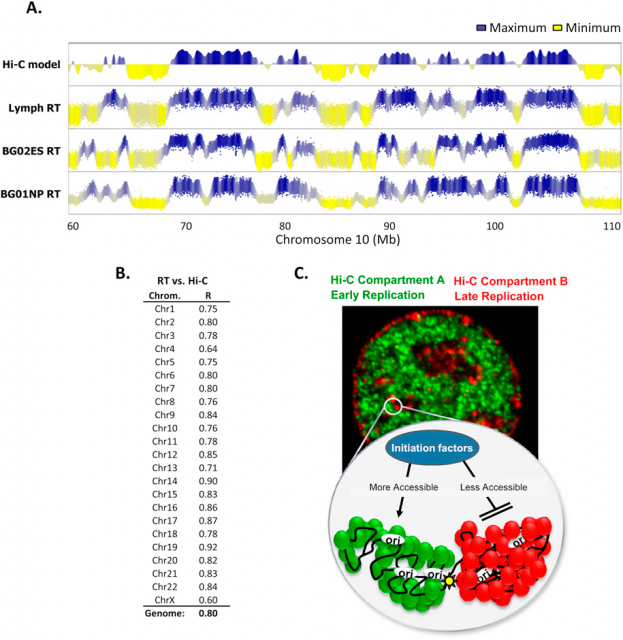
In the last decades many genome features have been related with the regulation of the replication-timing program. In fact, using the Hi-C (find more info about this technique here), It was observed that those genome regions that typically replicate simultaneously tend to be 3-dimensionally close (Fig.3). But not only the 3D structure of chromatin seems to be important. Former studies correlated the early DNA replication initiation with different genomic landmarks: GC-rich regions, regions rich in genes, G4-quadruplexes (guanine rich sequences capable of forming a four-stranded structure) and transcriptionally active regions (referred as transcription start sites: TSS).
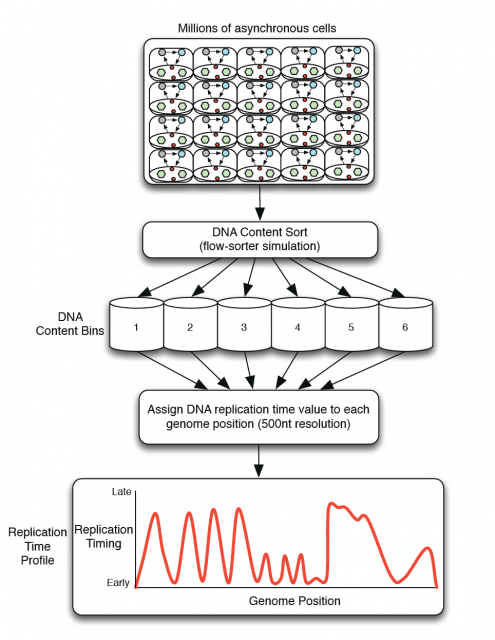
Amid the growing complexity of the mathematical models with multiple adjustable parameters that try to explain the regulation of the observed replication-timing program, Yevgeniy Gindin and coworkers2 bring a reductionist modelling approach, which try to not only reproduce but also predict the dynamic replication timing program of human cells. Unlike previous mathematical models, also accurate and confirmed, which implemented a time-dependent parameter to avoid instability in the S-phase duration; Gindin´s reductionist approach uses a time-independent input giving remarkably similar readouts. In this very recent paper, the mechanistic model proposed includes only two inputs of biological relevance. The main parameter is the IPLS (initiation probability landscape), a scaled value of an attribute of the genomic landmark that locates the initiation site. The other parameter is the number of replication forks (N). Basically it is a simulation that works as follows (Fig. 4A): Simulated cells can be either in a non-replicating or in a replicating state and the transition from the first to the second occurs randomly. When a virtual cell is in replicative state, the replication forks randomly choose a genome region (according with the landmark of choose in the model) and binds with a probability set by the IPLS. If the probability allows it to bind, replication goes on bi-directionally as far as its limited replication rate permit, restarting the process until the genome is replicated. Whenever the genome is replicated, the cell enters in a non-replicating state, repeating the process until simulation is terminated. Then, it is calculated the R-value as Pearson’s correlation between simulated and empirical data (Fig. 4B) and also the replication-time profile (e.g.: Fig. 4C). This mathematical model allows to test which of each different genome regions being involve in regulation of DNA replication initiation better fits experimentally observed data.
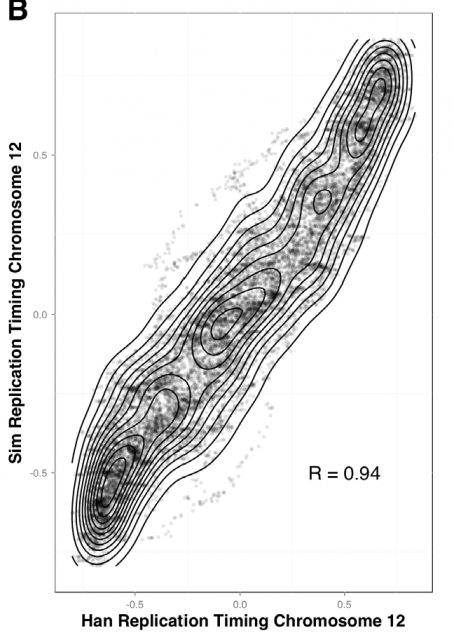
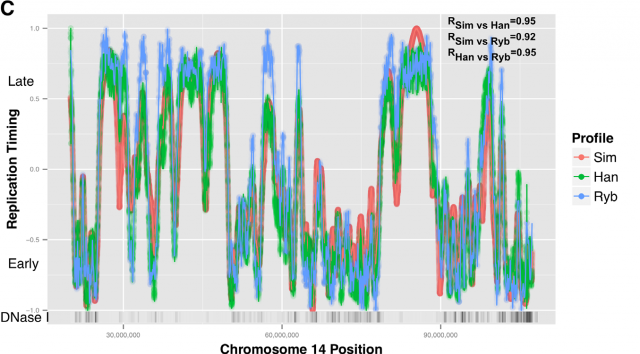
This way, Gindin et al. tested the predictive power of genomic features comparing the output of the modelling with the human replication timing data published by Hansen (Hansen et al, 2010) and (Ryba et al, 2012). By comparing different genomic landmarks like GC-rich regions, G4-quadruplexes and TSS with experimental data, they found that, even with the simplicity of this model, the prediction was rather good for TSS (average correlation: R=0,69) while a bit less predictive for the other features. In any case, the genomic features above are relatively similar within the human cells lineages under study and it seemed difficult that they could explain the replication-timing program flexibility observed across those cell types. Then, looking for more dynamic landmarks that could explain the timing plasticity, they made use of the data in ENCODE Project to generate new IPLSs. Form them; the best fitting IPLS (r = 0.87) is based on DNase-HS sites (Fig. 4B); which are genome regions with highly accessible DNA, hence being super sensible to cleavage by nucleases. Also good models were derived from IPLS obtained from other markers of active chromatin as H3k9ac (Histone 3 acetyl Lysine 9), H3k4me2 (Histone 3 dimethyl Lysine 4) or transcription factor binding regions. But in any case, only DNase-HS sites keep accuracy to predict in pairwise comparisons after subtraction of overlapping regions.
Given the potential limitation imposed for completeness of genomic annotations and a model with only one tuneable biological parameter as input, such a good fitting could point out the high degree of robustness in the system. To test if this was the case, they randomly removed up to 75% of the DNase-HS sites from the model without affect the level of prediction. Therefore, leading to one of the main general conclusions of this work and reinforcing what already suggested by previous models: the replication-timing program is an emergent phenomenon, so it does not depend on individual sites but on the collective contribution. Furthermore, they even alternate the probability assignment functions in the IPLS obtaining not significant changes in the prediction. The last gives away a second important point: the main player controlling the replication-timing program is the locations were PreRC assemblies (Fig. 2) but not the replication initiation by a bidirectional replication fork. In fact, once the appropriate landmark was found (in this case Dnase-HS), the timing could be predicted in other metazoan cells lines after extracting the landmark attribute from available genome data bases. Even the aberrant cycle displayed for a particular lymphoblastic cell line was reproduced after introducing in the model the redistribution of DNase-HS sites due to the chromosome translocation that characterize them.
All that seems to strengthen the idea that the initiation firing sequence, besides being a stochastic process, could gain its global accuracy, plasticity and regulation from the structural typology that emerges from the arrangement needed for the genes to make their job. Therefore, under consideration, which would be an unexpected emergent phenomenon, something that is not the sum of its parts. Hence, it would have been difficult to detect it using classical wet science without help of mathematical modelling applied to data obtained by high-throughput techniques. The mystery about how live cells are able to copy a genome of almost three thousand Megabases in few hours, with a strict but also adjustable temporal program, is being uncovered. To know its emergent nature, apart from shedding light on the molecular mechanistic of the process, will certainly be a shortcut to understand the behaviour of many diseases resulting from aberrant cell proliferation.
References
- Donley, N. & Thayer, M. J. DNA replication timing, genome stability and cancer: late and/or delayed DNA replication timing is associated with increased genomic instability. Semin. Cancer Biol.23, 80–9 (2013) ↩
- Gindin Y., Valenzuela M.S., Aladjem M.I., Meltzer P.S. & Bilke S. (2014). A chromatin structure-based model accurately predicts DNA replication timing in human cells, Molecular Systems Biology, 10 (3) 722-722. DOI: 10.1002/msb.134859 ↩
1 comment
[…] harrigarria da. Ikuskizun natural eder honetako sarrera txikia ematen digu Daniel Moreno Andrések DNA replication: how a job is well done in just a few hours […]