Bessel beam plane illumination microscopy: another smart solution for an old challenge.
Bessel beam plane illumination microscopy: another smart solution for an old challenge.
Since the emergence of the microscope in the early seventeenth century, many claimed its invention, but many more have tried to improve it. Many problems have been resolved on the way, allowing us to poke directly with our own eyes about the heart of the living or inert matter as far as optical physics allows it. After lenses refinement almost touched its theoretical zenith and hosts of fluorescent molecular markers have illuminated the darkness of the micro-world, there are still several limitations when it comes to observe living cells in all its glory. To film the journey of a cell is not easy. This required and requires a balance, since lots of light could generate toxic derivatives among the cell molecules (phototoxicity) or completely squeeze the brightness of the marker (bleaching). Just that: phototoxicity, low signal to background ratio and bleaching are the nightmares of all those wishing to extract the maximum of spatiotemporal information of the living micro-world.
Fluorescence microscopy emerged not long ago as the touchstone for the direct study of the inner life of the cell. The possibility of making visible the location and movement of structures and proteins and other molecules using different fluorescent markers renewed microscopy almost from its foundation: wide-field microscopes coupled fluorescent lamps to brighten fluorescent markers. However, out-of-focus light from the entire sample only let the observation of certain large and bright structures but not from those smaller and fainter. That was solved using special kind of collimator, called pinhole and developed by Marvin Minsky in the midle 50s. The pinhole is able to select only light in the focal plane increasing signal to background ratio, therefore the resolution, but also decreasing the brightness, thus making necessary to put more light in the sample. Nowadays, confocal microscopes became more sophisticated. They use lasers as light source and collimate the laser beams coordinately with the pinhole, allowing focusing the lighting beam to scan the sample instead to illuminate at once the whole specimen (Fig.1). By mean of scanning, confocal microscopes are able to build the image point-by-point (Rastrer imaging), with an increasingly better balance: less light, less toxicity and better signal and resolution.

But it is never good enough, reached an edge, it is needed to go beyond, that is the challenge. In this case, Thomas A. Planchon and coworkers 1 adapted a rather different microscopy technic called plane-illumination microscopy to achieve the greatest spatiotemporal resolution of a living cell up to date. Unlike a confocal, where the laser beam follows the same path than the acquired light, going through the entire sample during scanning, in this kind of light-sheet microscopy, the orthogonal lens disposition allows to expose the sample only in the plane of interest, performing the scanning during acquisition. Light-sheet microscopyis mainly used to image large multicellular organisms and its main advantage compared with confocal, when imaged big specimens, is less photo-bleaching and less out of focus light. (Fig. 2).
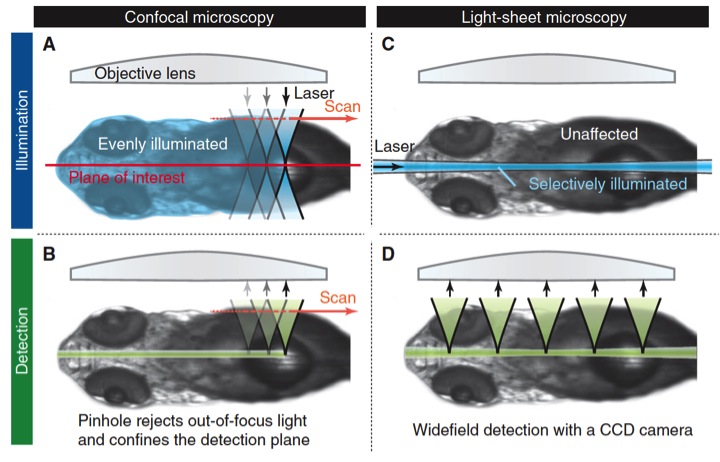
However, plane-illumination microscopy has a major disadvantage when being used with specimens smaller than 50 micrometers as isolated cells in culture. The light sheet used here is a Gaussian beam, which even optimized, is relatively divergent (spreads out after being focussed in to a small spot) and too thick for image such small sample without produce photobleaching and out-of-focus light. To face this drawback, Planchon and colleges replaced the Gaussian beam by a Bessel beam. Bessel beams are non-diffractive, so they do not spread out while propagate. And they have also a fascinating self-reconstruction property, which means that the beam is able recover its initial intensity profile after being partially obstructed by an obstacle.
The so called Bessel beam structured plane illumination microscopy uses wide-field acquisition and various modes of structured illumination with 3 and 9 phases and also a variant involving two photon illumination. By mean of those technical implementations it reduces the influence of the Bessel beam side lobes in the axial PSF (point-spread-function) (Fig. 3).
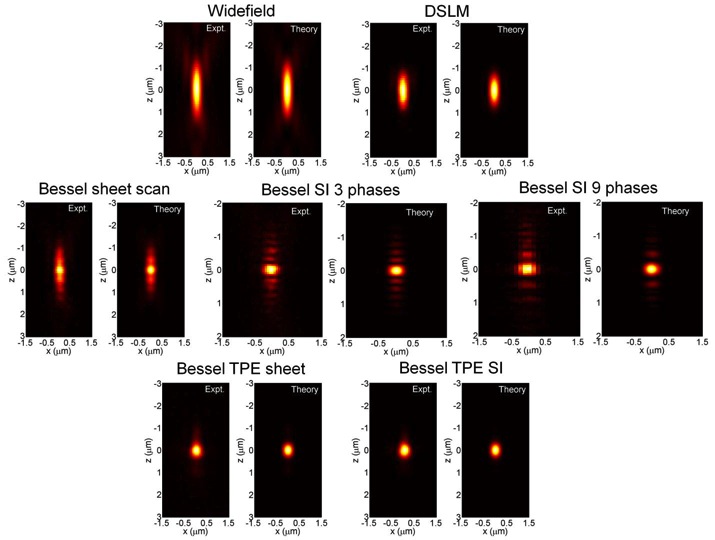
The application of these successive improvements results in an appreciable increase in the resolution after raw data deconvolution with the appropriate PSF in either case (Fig. 4 Left). Even more, appears to be comparatively better than other widely used confocal and light sheet fluorescence microscopy techniques like DSKM, showing that SI3 and TPE Bessel modes reduce out-of-focus light while having superior axial resolution and less photobleaching (Fig. 4 Right).
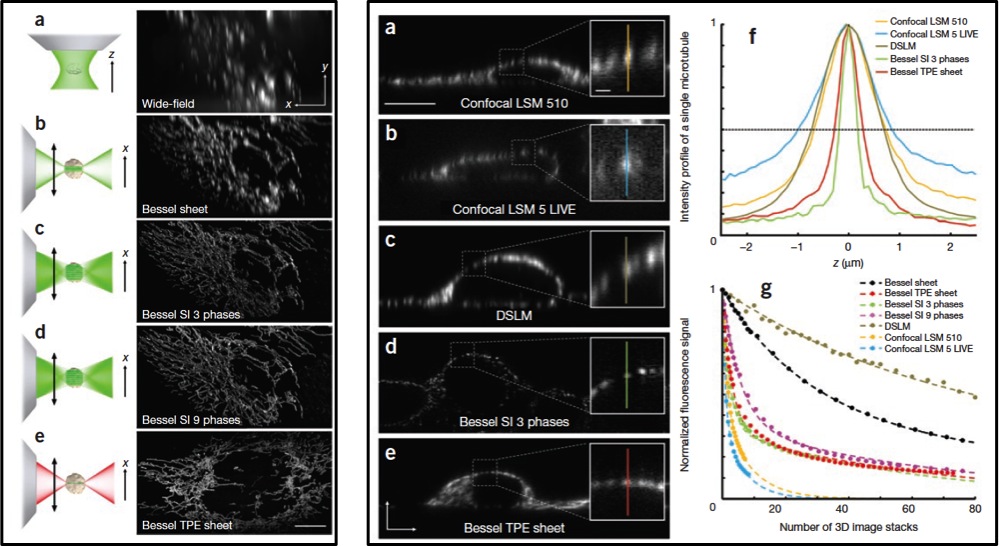
The reader has probably noticed that despite the introduction of this article, the capabilities of the Bessel beam structured plane illumination microscopy have been shown so far only for fixed samples. Nothing about living cells in all its glory. Needless to say that a video is worth than thousand images, more if it is to illustrate that all the technical improvements introduced above. Bessel beam structured plane illumination microscopy is able to create light sheets sufficiently thin and fast enough to achieve isotropic 3D resolution, pushing down the bleaching to an edge that allows recording hundreds of 3D image stacks in few seconds without allowing light needed to that to induce a phototoxicity level unbearable for Cell. A single living cell can be seen at 200 frames per second, uncovering its true nature as it had not been seen before (Videos 1, 2 and 3). In my opinion, this valuable approach will not only delight our eyes revealing the life of the cell as documentaries with life in the Savannah do, it opens a new dimension for the study of in vivo molecular dynamics where speed and resolution in measurements are paramount.
Video1 Dynamics of membrane ruffling (top) and intracellular vesicle motion (bottom) in a cSrc-transfected COS-7 cell.
Video2 Dynamics of membrane ruffling (top) and intracellular vesicle motion (bottom) in a cSrc-transfected COS-7 cell.
Video3 Fragmentation and reconstitution of the Golgi apparatus (magenta) during mitosis in a LLC-PK1 cell, after three-dimensional segmentation of single color Golgi and histone (green) data.
General reference: Optical Microscopy Primer
References
- Planchon, T. A. et al. Rapid three-dimensional isotropic imaging of living cells using Bessel beam plane illumination. Nat. Methods 8, 417–423 (2011) ↩