Inhibition of Hypoxia-Inducible Factor Alpha (HIF-1α) helps suppress T-ALL drug resistance
Inhibition of Hypoxia-Inducible Factor Alpha (HIF-1α) helps suppress T-ALL drug resistance
Hypoxia
Author: Marta Irigoyen is a postdoctoral researcher at CIC bioGUNE
Despite the fact that cancer treatments have greatly improved during recent years, chemoresistance remains a major problem in eradicating cancer cells. Drug resistance involves not only many cell intrinsic mechanisms but also extrinsic induced chemoprotection by the tumor microenvironment 1. In fact, this resistance may rely at least partly on less proliferating or even quiescent tumor cells escaping proliferating cell targeting drugs and preserving the ability to reinitiate cancers 2. Thus, identifying and resolving resistance mechanisms are currently major challenges in cancer treatment.
T cell acute lymphoblastic leukemia (T-ALL) is an aggressive hematologic malignancy characterized by bone marrow (BM) infiltration of immature lymphoblasts derived from transformed T-cell precursors 3. After chemotherapy, a small subpopulation of drug-resistant blasts survive below the detection limit making impossible the complete remission and increasing the probability that they will subsequently be reactivated, initiate again cell proliferation and trigger the relapse. However, the mechanisms underlying T-ALL chemoresistance are partially understood.
BM has been suggested to take part in a complex crosstalk in which leukemic cells can remodel the microenvironment 4. Hence, leukemia-supportive niches targeting treatments unveil a crucial aspect of T-ALL therapy 5. Hypoxia is a key microenvironmental status of BM that influences both the biology of hematopoietic stem cells and leukemic cells 6. In hypoxia, adaptive responses have been identified as downstream effects of hypoxia-inducible factors (HIFs) 7, being HIF-1 the master transcription factor for oxygen sensing. Since clinical investigations revealed that elevation of HIF-1α makes tumor cells more resistant to chemotherapy and increases metastasis and poor outcome 8, Lucine Fahy and collaborators investigated how low oxygen concentrations impact T-ALL proliferation, leukemia-propagating activity, and chemoresistance 9.
To perform this objective, authors firstly maintained human T-ALL cells for 4 days in 1% (hypoxia) and 21% O2 (hereafter called normoxia). Thus, they observed that hypoxia strongly inhibited T-ALL cell growth. Further analyzes showed that hypoxia, rather than inducing apoptosis, impacted T-ALL growth by increasing quiescence and reducing cell cycle progression. As cell exposure to hypoxic conditions may also alter energetic metabolism, they incubated T-ALL with two different fluorochromes to assess mitochondrial content and mitochondrial membrane potential. They detected low levels of fluorochromes signals in T-ALL cells under hypoxia compared with normoxic environment, indicating that mitochondrial mass decrease and provokes a weaker activity or depolarization of the mitochondria membrane. Besides, increased anaerobic glycolysis was observed with elevation of lactate levels in T-ALL culture medium as O2 levels decreased, further indicative of metabolic switch in hypoxia. Altogether, these data suggest an increased quiescence and a significant mitochondrial rewiring in leukemic cells under hypoxia.
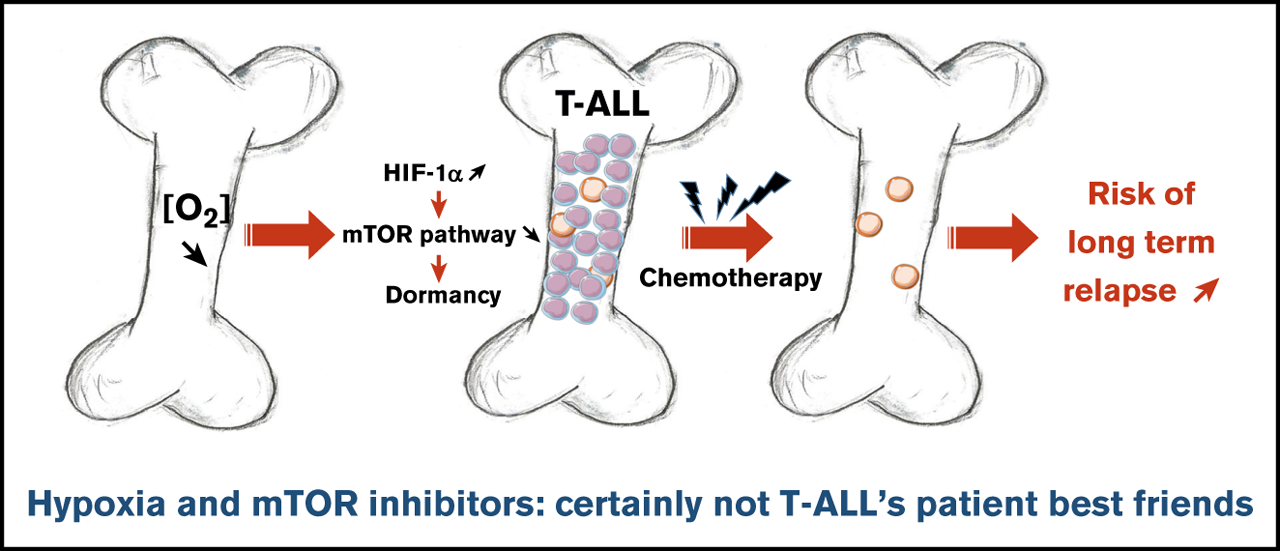
Next, the authors assumed that the intrinsic T-ALL drugs sensitivity might differ whether the leukemic cells were cultured in hypoxia or normoxia. To test this hypothesis, leukemic cells were preincubated 24 hours in hypoxia or normoxia and then treated for an additional 72 hours in medium containing vincristine sulfate, cytarabine, or dexamethasone while maintaining respective O2 tension. By calculating the percentage of live cells after drug exposure, they further observed that hypoxic T-ALL cells displayed a lower sensitivity to different drugs compared with normoxic cells. Besides, cells treated with vincristine in hypoxia before graft in immune-deficient mice generated leukemia that developed faster in the mouse BM and animals died of leukemia 4 times faster than the mice treated with vincristine in normoxia. These results indicated that hypoxia lowers T-ALL sensitivity to chemotherapy and provides a resistance status.
Hypoxia has been shown to promote cancer stemness through HIF factors 10. Since HIF-2α transcripts are weakly expressed in T-ALL, authors focused their work on HIF-1α and knocked down (KD) its expression to evaluate its role in hypoxia-mediated growth, quiescence and chemoresistance. Interestingly, the growth of T-ALL cells that is decreased in hypoxia was partially restored on HIF-1α KD. Furthermore, when transduced T-ALL cells were cultured in hypoxia, the proportion of quiescent leukemic cells was lower in HIF-1α KD compared with the control condition. These results indicated that HIF-1α silencing is able to decreases hypoxia-induced cell quiescence.
Next, they used HIF-1α KD T-ALL blasts to assess whether HIF-1α inhibition could modify hypoxia-induced drug resistance. They observed that silencing HIF-1α in T-ALL blocked the hypoxia-mediated resistance to vincristine, reaching same levels of live cells as T-ALL cells treated with vincristine in normoxia. These results indicate that HIF-1α is a major effector in the hypoxia-related drug resistance of T-ALL. They next sought to determine whether HIF-1α levels also impacted T-ALL chemosensitivity in vivo. Leukemia propagation was studied using a (1:1) competitive transplantation of shHIF-1α (GFP+) cells mixed with shCTL (Cherry+) cells from T-ALL and mice were treated for a week with a combination of vincristine, l-asparaginase, dexamethasone, and cytarabine 11. Importantly they observed a significant drop of shHIF-1α T-ALL cells and inversely an increase of shCTL cells in the treated mouse BM. These results indicate that HIF-1α silencing sensitizes T-ALL to treatment, further pointing out the importance of HIF-1α levels for T-ALL chemoresistance in vivo.
In response to hypoxia, cells rapidly activate a variety of adaptive mechanisms that limit energy expenditure through the inhibition of energy-intensive processes including protein translation 12. Hence, a major mechanism implicates that mTOR activity is inhibited on hypoxia exposure (7). In accordance, activation of the mTOR pathway was decreased in T-ALL cells in hypoxia. Interestingly, treating T-ALL with rapamycin (Rapa), an inhibitor of the mTORC1 protein kinase, mimicked the effects of hypoxia. Indeed, Rapa inhibited in vitro leukemic cell growth in normoxia. Also, T-ALL blasts pretreated for 45 minutes with Rapa before an additional 3 days of anticancer drug treatment displayed a lower sensitivity to vincristine, cytarabine, or dexamethasone in normoxia. Interestingly, the inhibition of mTOR activation mediated by hypoxia was abolished in HIF-1α KD T-ALL cells. Altogether, these results reveal that mTOR and HIF-1α pathways interact and participate in hypoxia-mediated drug resistance in T-ALL.
In summary, authors demonstrate that hypoxic niches play a protective role of T-ALL during treatments. Inhibition of HIF-1α and activation of the mTORC1 pathway may help suppress the drug resistance of T-ALL in hypoxic niches. Hence, the use of mTORC1 inhibitors for T-ALL patient treatment may have long-term adverse effects, with a potentially increased risk of relapse from quiescent chemoresistant leukemic cells.
References
- Taylor ST, Hickman JA, Dive C. Epigenetic determinants of resistance to etoposide regulation of Bcl-X(L) and Bax by tumor microenvironmental factors. J Natl Cancer Inst 2000; 92: 12-23. PMID: 10620629. doi: 10.1093/jnci/92.1.18. ↩
- Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer 2014;14: 611-22. PMID: 25118602 DOI: 10.1038/nrc3793. ↩
- Pui CH, Robinson LL, Look AT. Acute lymphoblastic leukaemia. Lancet 2008; 371: 1030-1043. PMID: 18358930 DOI: 10.1016/S0140-6736(08)60457-2. ↩
- Schepers K, Pietras EM, Reynaud D, Flach R, Binnewies M, Garg T et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 2013; 13: 285-299. PMID: 23850243 DOI: 10.1016/j.stem.2013.06.009. ↩
- Weisberg E, Azab AK, Manley PW, Kung AL, Christie AL, Bronson R et al.. Inhibition of CXCR4 in CML cells disrupts their interaction with the bone marrow microenvironment and sensitizes them to nilotinib. Leukemia 2012; 26: 985-990. PMID: 22182920 DOI: 10.1038/leu.2011.360. ↩
- Forristal CE, Winkler IG, Nowlan B, Barbier V, Walkinshaw G, Levesque JP. Pharmacologic stabilization of HIF-1α increases hematopoietic stem cell quiescence in vivo and accelerates blood recovery after severe irradiation. Blood 2013; 121: 759-769. PMID: 23243286 DOI: 10.1182/blood-2012-02-408419. ↩
- Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer 2003; 3721-732. PMID: 13130303 DOI: 10.1038/nrc1187. ↩
- Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer 2004; 4: 437-447. PMID: 15170446 DOI: 10.1038/nrc1367. ↩
- Fahy L, Calvo J, Chabi S, Renou L, Le Maout C, Poglio S et al. Hypoxia favors chemoresistance in T-ALL through an HIF1α-mediated mTORC1 inhibition loop. Blood 2021; 5: 513–526. PMID: 33496749 DOI: 10.1182/bloodadvances.2020002832. ↩
- Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev 2007; 26: 225-239. PMID: 17440684 DOI: 10.1007/s10555-007-9055-1. ↩
- Samuels AL, Beesley AH, Yadav BD, Papa RA, Sutton R, Anderson A et al. A pre-clinical model of resistance to induction therapy in pediatric acute lymphoblastic leukemia. Blood Cancer J 2014; 4: e232. PMID: 25083816 DOI: 10.1038/bcj.2014.52. ↩
- Masoud GN, Li W. HIF-1α pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B 2015; 5: 378-389. PMID: 26579469 DOI: 10.1016/j.apsb.2015.05.007. ↩